









![一种不对称[3+2]环化反应合成手性五元碳环嘌呤核苷的方法](https://www.zhichawang.com/images/ui/CN2017109038084/CN2017109038084.jpg)
专利摘要
酯化纤维素醚适用于制备包含药物的固体分散体,所述酯化纤维素醚包含(i)脂肪族单价酰基或(ii)式-C(O)-R-COOA的基团,其中R是二价脂肪族或芳香族烃基,并且A是氢或阳离子,或(iii)脂肪族单价酰基与所述式-C(O)-R-COOA的基团的组合,所述酯化纤维素醚在20℃下以10wt%酯化纤维素醚的丙酮溶液形式测量的粘度是至多19mPa·s,并且其重量平均分子量Mw是至少90,000道尔顿。
权利要求
1.一种酯化纤维素醚,
其包含(i)脂肪族单价酰基;或(ii)式-C(O)-R-COOA的基团,其中R是二价脂肪族或芳香族烃基,并且A是氢或阳离子,或(iii)脂肪族单价酰基与所述式-C(O)-R-COOA的基团的组合,
所述酯化纤维素醚在20℃下以10wt%所述酯化纤维素醚的丙酮溶液测量的粘度是至多19mPa·s,并且
其重量平均分子量Mw是至少90,000道尔顿。
2.根据权利要求1所述的酯化纤维素醚,另外其在20℃下以2.0wt%所述酯化纤维素醚在0.43wt%NaOH水溶液中的溶液形式测量的粘度是至多2.33mPa·s。
3.根据权利要求1或2所述的酯化纤维素醚,其重量平均分子量Mw是90,000到220,000道尔顿,并且在20℃下以10wt%所述酯化纤维素醚的丙酮溶液形式测量的粘度是1.50mPa·s到15mPa·s。
4.根据权利要求1或2所述的酯化纤维素醚,其重量平均分子量Mw是220,000到350,000道尔顿,并且在20℃下以10wt%所述酯化纤维素醚的丙酮溶液形式测量的粘度是5到18mPa·s。
5.根据权利要求1到4中任一权利要求所述的酯化纤维素醚,其中所述脂肪族单价酰基是乙酰基、丙酰基或丁酰基,并且所述式-C(O)-R-COOA的基团是-C(O)-CH2-CH2-COOA、-C(O)-CH=CH-COOA或-C(O)-C6H4-COOA基团。
6.根据权利要求1到5中任一权利要求所述的酯化纤维素醚,其为酯化羟基烷基甲基纤维素。
7.根据权利要求1到6中任一权利要求所述的酯化纤维素醚,其为羟基丙基甲基纤维素乙酸酯丁二酸酯。
8.一种组合物,其包含液体稀释剂和至少一种根据权利要求1到7中任一权利要求所述的酯化纤维素醚。
9.根据权利要求8所述的组合物,其另外包含至少一种活性成分和任选地一或多种佐剂。
10.根据权利要求8或9所述的组合物,其包含以所述组合物的总重量计10到25%的至少一种酯化纤维素醚、70到89%的液体稀释剂以及1到15%的活性成分。
11.一种固体分散体,其包含至少一种活性成分和至少一种根据权利要求1到7中任一权利要求所述的酯化纤维素醚。
12.根据权利要求11所述的固体分散体,其中所述固体分散体配制成片剂、丸剂、颗粒、丸粒、囊片、微粒、胶囊的填充物或糊剂、乳膏、悬浮液或浆液。
13.一种剂型,其涂布有至少一种根据权利要求1到7中任一权利要求所述的酯化纤维素醚。
14.一种胶囊壳,其包含至少一种根据权利要求1到7中任一权利要求所述的酯化纤维素醚。
15.一种制备根据权利要求1到7中任一权利要求所述的酯化纤维素醚的方法,其中用(i)脂肪族单羧酸酐或(ii)二羧酸酐或(iii)脂肪族单羧酸酐与二羧酸酐的组合酯化在20℃下以2wt.-%水溶液形式测量粘度是1.20到2.33mPa·s的纤维素醚。
说明书
技术领域
本发明涉及新颖酯化纤维素醚、活性成分在此类酯化纤维素醚中的固体分散体,以及包含此类酯化纤维素醚的液体组合物、经涂布剂型和胶囊。
背景技术
纤维素醚的酯、其用途和其制备方法在所属领域中是一般已知的。制备纤维素醚-酯的已知方法包括使纤维素醚与脂肪族单羧酸酐或二羧酸酐或其组合反应,例如如美国专利第4,226,981号和第4,365,060号中所述。
各种已知的酯化纤维素醚适用作医药剂型的肠溶聚合物,如甲基纤维素邻苯二甲酸酯、羟基丙基甲基纤维素邻苯二甲酸酯、甲基纤维素丁二酸酯或羟基丙基甲基纤维素丁二酸酯。涂布有此类聚合物的剂型避免药物在酸性环境中失活或降解或防止药物刺激胃。美国专利第4,365,060号公开据称具有极佳肠溶性性能的肠溶性胶囊。
美国专利申请公开案第2004/0152886号公开羟基丙基甲基纤维素邻苯二甲酸酯的制备,其用以2wt.%水溶液形式测量粘度为3到20cp的羟基丙基甲基纤维素作为起始物质。
国际专利申请案WO 2005/115330和WO 2011/159626公开羟基丙基甲基纤维素乙酸酯丁二酸酯(HPMCAS)的制备。推荐表观粘度为2.4到3.6cp的HPMC作为起始物质。或者,推荐600到60,000道尔顿、优选3000到50,000道尔顿、更优选6,000到30,000道尔顿的HPMC起始物质。根据基尔里(Keary)[基尔里,C.M.;碳水化合物聚合物45(2001)293-303(Keary,C.M.;Carbohydrate Polymers 45(2001)293-303),表7和表8],重量平均分子量为约85到100kDa的HPMC以2重量%水溶液形式测定的粘度是约50mPa×s。
美国专利第5,776,501号教示水溶性纤维素醚的用法,所述水溶性纤维素醚以2重量%水溶液形式测定粘度是3到10cp(mPa·s)。如果粘度小于3cp,那么最终所得的用于固体肠溶药物制剂的涂层膜强度不足,而如果其超过10cp,那么在将其溶解在溶剂中以执行取代反应时观察到的粘度变得极高。
欧洲专利申请EP-A-0 219 426公开一种制备纤维素醚的肠溶性酸性二羧酸酯的方法,所述二羧酸酯由在20℃下以2重量%水溶液形式测量粘度为至少5cp的纤维素醚制成。与在拟胃液中崩解的由3cp粘度的HPMC制成的HPMCAS相比,由6cp粘度的HPMC制成的HPMCAS具有较高分子量和对拟胃液的良好抗性。
尽管高分子量酯化纤维素醚极适宜用于对胃液产生良好抗性,但已知高分子量酯化纤维素醚在其以高浓度(如7-10重量%的浓度)溶解于有机溶剂中时展现高粘度,从而降低其在涂布过程中的效率。在涂布过程中,需要高浓度的酯化纤维素醚在有机溶剂中的溶液以使随后必须移除的溶剂的量达最少。另一方面,溶液的粘度应低到可有利于在欲涂布的剂型(如片剂)上喷洒溶液。
此外,大量当前已知药物在水中具有低溶解性,使得需要复杂技术来制备剂型。一种已知方法包括将此类药物与药物学上可接受的水溶性聚合物(如酯化纤维素醚)一起溶解于任选地与水掺合的有机溶剂中,且喷雾干燥所述溶液。酯化纤维素醚旨在降低药物的结晶度,从而使药物溶解所要的活化能达最小,以及在药物分子周围建立亲水性条件,从而改良药物自身的溶解性以提高其生物可用性,即,摄取后个体对其的活体内吸收。在喷雾干燥过程中,也需要高浓度的酯化纤维素醚在有机溶剂中的溶液以使必须移除的溶剂的量达最少。但是,当包含酯化纤维素醚的有机溶液的粘度高时,溶液难以喷雾干燥并且趋向于堵塞喷雾干燥装置。
因此,高度需要寻找到新颖酯化纤维素醚,其具有高分子量但其仍可以有效用于喷雾干燥和涂布过程。
发明内容
本发明的一个方面是一种酯化纤维素醚,其包含(i)脂肪族单价酰基,或(ii)式-C(O)-R-COOA的基团,其中R是二价脂肪族或芳香族烃基,并且A是氢或阳离子,或(iii)脂肪族单价酰基与式-C(O)-R-COOA的基团的组合,所述酯化纤维素醚在20℃下以10wt%酯化纤维素醚的丙酮溶液形式测量粘度是至多19mPa·s,并且其重量平均分子量Mw是至少90,000道尔顿。
本发明的另一方面是一种包含液体稀释剂和至少一种上述酯化纤维素醚的组合物。
本发明的另一方面是至少一种活性成分在至少一种上述酯化纤维素醚中的一种固体分散体。
本发明的另一方面是一种剂型,其涂布有至少一种上述酯化纤维素醚。
本发明的另一方面是一种包含至少一种上述酯化纤维素醚的胶囊壳。
本发明的另一方面是一种制备上述酯化纤维素醚的方法,其中用(i)脂肪族单羧酸酐或(ii)二羧酸酐或(iii)脂肪族单羧酸酐与二羧酸酐的组合酯化纤维素醚,所述纤维素醚在20℃下以2wt-%水溶液形式测量粘度是1.20到2.33mPa·s。
具体实施方式
酯化纤维素醚具有如下纤维素主链,其具有在本发明的上下文中称为脱水葡萄糖单元的β-1,4糖苷结合的D-吡喃葡萄糖重复单元。酯化纤维素醚优选是酯化烷基纤维素、羟基烷基纤维素或羟基烷基烷基纤维素。这意味着在本发明的酯化纤维素醚中,脱水葡萄糖单元的羟基的至少一部分经烷氧基或羟基烷氧基或烷氧基与羟基烷氧基的组合取代。羟基烷氧基通常是羟基甲氧基、羟基乙氧基和/或羟基丙氧基。羟基乙氧基和/或羟基丙氧基是优选的。通常一或两种类别的羟基烷氧基存在于酯化纤维素醚中。优选存在单一类别的羟基烷氧基,更优选羟基丙氧基。烷氧基通常是甲氧基、乙氧基和/或丙氧基。甲氧基是优选的。以上所定义的酯化纤维素醚的说明性实例是酯化烷基纤维素,如酯化甲基纤维素、乙基纤维素和丙基纤维素;酯化羟基烷基纤维素,如酯化羟基乙基纤维素、羟基丙基纤维素和羟基丁基纤维素;以及酯化羟基烷基烷基纤维素,如酯化羟基乙基甲基纤维素、羟基甲基乙基纤维素、乙基羟基乙基纤维素、羟基丙基甲基纤维素、羟基丙基乙基纤维素、羟基丁基甲基纤维素和羟基丁基乙基纤维素;以及具有两个或两个以上羟基烷基的物质,如酯化羟基乙基羟基丙基甲基纤维素。最优选地,酯化纤维素醚是酯化羟基烷基甲基纤维素,如羟基丙基甲基纤维素。
脱水葡萄糖单元的羟基经羟基烷氧基取代的程度通过羟基烷氧基的摩尔取代度MS(羟基烷氧基)表示。MS(羟基烷氧基)是酯化纤维素醚中每个脱水葡萄糖单元的羟基烷氧基摩尔数的平均数。应了解,在羟基烷基化反应期间,结合到纤维素主链的羟基烷氧基的羟基可进一步通过烷基化剂(例如甲基化剂)和/或羟基烷基化剂醚化。对于脱水葡萄糖单元的同一碳原子位置的多个连续羟烷基化醚化反应产生侧链,其中多个羟基烷氧基通过醚键彼此共价结合,每个侧链作为整体形成纤维素主链的羟基烷氧基取代基。
因此,在MS(羟烷氧基)的情况下在提到羟基烷氧基时,术语“羟基烷氧基”必须解释为羟基烷氧基取代基的构成单元,其包含单一羟基烷氧基或如上所述的侧链,其中两个或两个以上羟基烷氧基单元通过醚键结而彼此共价结合。在此定义内,羟基烷氧基取代基的末端羟基进一步烷基化或未烷基化并不重要;对于确定MS(羟烷氧基)包括烷基化与非烷基化的羟烷氧基取代基。本发明的酯化纤维素醚的羟基烷氧基摩尔取代度在0.05到1.00范围内,优选0.08到0.90、更优选0.12到0.70、最优选0.15到0.60并且尤其0.21到0.50范围内。
每个脱水葡萄糖单元经烷氧基(如甲氧基)取代的羟基的平均数称为烷氧基的取代度,即DS(烷氧基)。在上文给出的DS的定义中,术语“经烷氧基取代的羟基”在本发明内解释为不仅包括直接结合到纤维素主链的碳原子的烷基化羟基,而且包括结合到纤维素主链的羟烷氧基取代基的烷基化羟基。根据本发明的酯化纤维素醚的DS(烷氧基)优选在1.0到2.5范围内,更优选是1.1到2.4,甚至更优选1.2到2.2,最优选1.6到2.05,并且尤其1.7到2.05。
酯化纤维素醚最优选是酯化羟基丙基甲基纤维素,对于DS(烷氧基),其DS(甲氧基)在上文指定的范围内,并且对于MS(羟基烷氧基),其MS(羟基丙氧基)在上文指定的范围内。
本发明的酯化纤维素醚具有(i)脂肪族单价酰基,或(ii)式-C(O)-R-COOA的基团,其中R是二价脂肪族或芳香族烃基,并且A是氢或阳离子,或(iii)脂肪族单价酰基与式-C(O)-R-COOA的基团的组合。阳离子优选是铵阳离子,如NH4,或碱金属离子,如钠或钾离子,更优选钠离子。最优选地,A是氢。
脂肪族单价酰基优选选自由以下基团组成的群组:乙酰基、丙酰基以及丁酰基,如正丁酰基或异丁酰基。
优选的式-C(O)-R-COOA的基团是-C(O)-CH2-CH2-COOA,如-C(O)-CH2-CH2-COOH或-C(O)-CH2-CH2-COO-Na+,
-C(O)-CH=CH-COOA,如-C(O)-CH=CH-COOH或-C(O)-CH=CH-COO-Na+,或
-C(O)-C6H4-COOA,如-C(O)-C6H4-COOH或-C(O)-C6H4-COO-Na+。
在式-C(O)-C6H4-COOA的基团中,羰基和羧酸基团优选以邻位排列。
优选的酯化纤维素醚是
i)HPMCXY,其中HPMC是羟基丙基甲基纤维素,X是A(乙酸酯),或X是B(丁酸酯)或X是Pr(丙酸酯),并且Y是S(丁二酸酯),或Y是P(邻苯二甲酸酯)或Y是M(顺丁烯二酸酯),如羟基丙基甲基纤维素乙酸酯邻苯二甲酸酯(HPMCAP)、羟基丙基甲基纤维素乙酸酯顺丁烯二酸酯(HPMCAM)或羟基丙基甲基纤维素乙酸酯丁二酸酯(HPMCAS),或
ii)羟基丙基甲基纤维素邻苯二甲酸酯(HPMCP)、羟基丙基纤维素乙酸酯丁二酸酯(HPCAS)、羟基丁基甲基纤维素丙酸酯丁二酸酯(HBMCPrS)、羟基乙基羟基丙基纤维素丙酸酯丁二酸酯(HEHPCPrS);以及甲基纤维素乙酸酯丁二酸酯(MCAS)。
羟基丙基甲基纤维素乙酸酯丁二酸酯(HPMCAS)是最优选的酯化纤维素醚。
酯化纤维素醚的脂肪族单价酰基(如乙酰基、丙酰基或丁酰基)取代度通常是0到1.75,优选0.05到1.50,更优选0.10到1.25,并且最优选0.20到1.00。
酯化纤维素醚的式-C(O)-R-COOA的基团(如丁二酰基)的取代度是0.05到1.6,优选0.05到1.30,更优选0.05到1.00,并且最优选0.10到0.70或甚至0.10到0.60。
i)脂肪族单价酰基的取代度与ii)式-C(O)-R-COOA的基团的取代度的总和通常是0.05到2.0,优选0.10到1.4,更优选0.20到1.15,最优选0.30到1.10,并且尤其0.40到1.00。
乙酸酯和丁二酸酯酯基的含量根据“羟丙甲纤维素乙酸酯丁二酸酯”,《美国药典和国家处方集》,NF 29,第1548-1550页(“Hypromellose Acetate Succinate”,United States Pharmacopeia and National Formulary,NF 29,pp.1548-1550)确定。将报告值针对挥发物进行校正(如在上述HPMCAS专论中的章节“干燥失重”中所述确定)。所述方法可以类似方式用于确定丙酰基、丁酰基、苯二甲酰基和其它酯基的含量。
酯化纤维素醚中醚基的含量以如对于“羟丙甲纤维素”,《美国药典和国家处方集》,USP 35,第3467-3469页(“Hypromellose”,United States Pharmacopeia and National Formulary,USP 35,pp 3467-3469)所述相同的方式确定。
根据下式,将通过以上分析获得的醚和酯基的含量转化为个别取代基的DS和MS值。所述式可以类似方式用于确定其它纤维素醚酯的取代基的DS和MS。
M(MeO)=M(OCH3)=31.03Da M(HPO)=M(OCH2CH(OH)CH3)=75.09Da
M(乙酰基)=M(COCH3) M(丁二酰基)=M(COC2H4COOH)=101.08Da =43.04Da
M(AGU)=162.14Da M(OH)=17.008Da M(H)=1.008Da
按照惯例,重量百分比是以纤维素重复单元的总重量计(包括所有取代基)的平均重量百分比。甲氧基的含量根据甲氧基(即,-OCH3)的质量报告。羟基烷氧基的含量根据羟基烷氧基(即,-O-亚烷基-OH)的质量报告;如羟基丙氧基(即,-O-CH2CH(CH3)-OH)。脂肪族单价酰基的含量根据-C(O)-R1的质量报告,其中R1是单价脂族基,如乙酰基(-C(O)-CH3)。式-C(O)-R-COOH的基团的含量根据此基团的质量(如丁二酰基(即,-C(O)-CH2-CH2-COOH)的质量)报告。
本发明的酯化纤维素醚在20℃下以10wt%酯化纤维素醚的丙酮溶液形式测量粘度至多仅为19mPa·s,通常至多17mPa·s,并且在本发明的一些实施例中甚至至多仅为15mPa·s,更优选至多13mPa·s,并且甚至更优选至多11mPa·s。在本发明的一些实施例中,酯化纤维素醚可以经制备在20℃下以10wt%酯化纤维素醚的丙酮溶液形式测量粘度甚至仅为至多8.0mPa·s,优选仅为至多6.5mPa·s,更优选仅为至多5.0mPa·s,最优选仅为至多4.0mPa·s,并且尤其仅为至多3.0mPa·s。本发明的酯化纤维素醚在20℃下以10wt%酯化纤维素醚的丙酮溶液形式测量粘度通常是1.50mPa·s或1.50mPa·s以上,更通常1.65mPa·s或1.65mPa·s以上,并且最通常1.80mPa·s或1.80mPa·s以上。在本发明的一些实施例中,酯化纤维素醚的粘度是5.0mPa·s或5.0mPa·s以上,更通常6.5mPa·s或6.5mPa·s以上。以10wt%丙酮溶液测量,本发明的酯化纤维素醚的粘度显著低于已知酯化纤维素醚在丙酮中的粘度。当包含酯化纤维素醚的液体组合物进行喷雾干燥时,本发明的酯化纤维素醚的低粘度非常有利于例如制备包含活性成分和酯化纤维素醚的固体分散体。10wt%酯化纤维素醚的丙酮溶液可以如以下实例中进一步所述来制备。
本发明的酯化纤维素醚的平均分子量取决于各种因素,如在20℃下以2.0wt%水溶液形式测量的用于制备本发明的酯化纤维素醚的纤维素醚的粘度,以及用于酯化反应的纤维素醚、稀释剂和催化剂之间的重量比,其将在下文中更详细说明。
在本发明的一个实施例中,酯化纤维素醚在20℃下以2.0wt%酯化纤维素醚在0.43wt.-%NaOH水溶液中的溶液形式测量的粘度是至多2.33mPa·s,或至多2.25mPa·s,优选至多2.10mPa·s,更优选至多1.95mPa·s,甚至优选至多1.80mPa·s,最优选至多1.70mPa·s,并且尤其至多1.55mPa·s。在20℃下以2.0wt%酯化纤维素醚在0.43wt%NaOH水溶液中的溶液形式测量,酯化纤维素醚的粘度通常是1.20mPa·s或1.20mPa·s以上,通常1.30mPa·s或1.30mPa·s以上,并且更通常1.40mPa·s或1.40mPa·s以上。2.0重量%酯化纤维素醚溶液如“羟丙甲纤维素乙酸酯丁二酸酯”,《美国药典和国家处方集》,NF 29,第1548-1550页所述制备,接着根据DIN 51562-1:1999-01(1999年1月)进行乌氏粘度(Ubbelohde viscosity)测量。已发现酯化纤维素醚在0.43wt%NaOH水溶液中的粘度与适用作制备酯化纤维素醚的起始物质的纤维素醚的粘度极类似。
本发明的酯化纤维素醚的重量平均分子量Mw是至少90,000道尔顿,优选95,000到500,000道尔顿,更优选100,000到350,000道尔顿,并且尤其105,000到300,000道尔顿。
在一个实施例中,本发明的酯化纤维素醚的重量平均分子量Mw是90,000到220,000道尔顿,通常100,000到200,000道尔顿,并且更通常100,000到180,000道尔顿,并且在20℃下以10wt%酯化纤维素醚的丙酮溶液形式测量粘度是至多17mPa·s,通常1.50mPa·s到15mPa·s,更通常1.65到13mPa·s,并且最通常1.80mPa·s到11mPa·s。在另一实施例中,本发明的酯化纤维素醚的重量平均分子量是220,000到500,000道尔顿,通常230,000到350,000道尔顿,并且更通常250,000到300,000道尔顿,并且在20℃下以10wt%酯化纤维素醚的丙酮溶液形式测量粘度是至多19mPa·s,通常5到18mPa·s,并且更通常8到17mPa·s。当酯化纤维素醚的重量平均分子量增加时,其通常意味着其在丙酮中的粘度也增加。意外地,本发明的酯化纤维素醚具有所提及的高重量平均分子量以及上述在丙酮中的低粘度。
Mw、Mn和Mz根据《医药和生物医学分析杂志》,56(2011)743(Pharmaceutical and Biomedical Analysis 56(2011)743)使用40体积份乙腈和60体积份含有50mM NaH2PO4 和0.1M NaNO3的水性缓冲液的混合物作为流动相测量。将流动相调节到pH 8.0。Mw、Mn和Mz的测量更详细地描述在实例中。
以下实例描述如何制备本发明的酯化纤维素醚。制备这些酯化纤维素醚的方法的一些方面以如下较通用术语描述。
在制备本发明的酯化纤维素醚的优选方法中,用(i)脂肪族单羧酸酐或(ii)二羧酸酐或(iii)脂肪族单羧酸酐与二羧酸酐的组合酯化纤维素醚,所述纤维素醚在20℃(+/-0.1℃)下以2.0wt%水溶液形式测量的粘度是1.20到2.33mPa·s,通常1.20到2.25mPa·s,优选1.20到2.10mPa·s,更优选1.20到1.95mPa·s,甚至更优选1.20到1.80mPa·s,最优选1.20到1.70mPa·s,并且尤其1.20到1.55mPa·s。所述方法更详细地描述于下文中。
2.0重量%纤维素醚水溶液根据美国药典(United States Pharmacopeia)(USP 35,“羟丙甲纤维素(Hypromellose)”,第3467-3469页)制备,接着根据DIN 51562-1:1999-01(1999年1月)进行乌氏粘度测量。用作起始物质的低粘度纤维素醚使得可以充分混溶用于制备酯化纤维素醚的反应混合物,而得到均匀反应混合物。优选使用如下纤维素醚,其具有上文进一步描述的类型的醚基和醚基的取代度。上述纤维素醚和其制备描述于国际专利申请案WO2009061821和WO2009/061815中。
纤维素醚与(i)脂肪族单羧酸酐或(ii)二羧酸酐或(ii)脂肪族单羧酸酐与二羧酸酐的组合反应。优选的脂肪族单羧酸酐选自由以下物质组成的群组:乙酸酐、丁酸酐以及丙酸酐。优选的二羧酸酐选自由以下物质组成的群组:丁二酸酐、顺丁烯二酸酐以及邻苯二甲酸酐。如果组合使用二羧酸酐与脂肪族单羧酸酐,那么两种酸酐可以同时引入反应容器中或一个接一个地引入反应容器中。欲引入反应容器中的每种酸酐的量取决于最终产物中欲获得的所要酯化度,通常是脱水葡萄糖单元通过酯化产生的所要摩尔取代度的化学计量量的1到10倍。
如果使用二羧酸的酸酐,那么[二羧酸的酸酐/纤维素醚的脱水葡萄糖单元]的摩尔比通常是0.1/1到1.5/1,优选0.13/1到1/1,以制备重量平均分子量Mw是90,000到220,000道尔顿、通常100,000到200,000道尔顿并且更通常100,000到180,000道尔顿的酯化纤维素醚。如果使用二羧酸的酸酐,那么[二羧酸的酸酐/纤维素醚的脱水葡萄糖单元]的摩尔比通常是0.3/1到0.4/1,优选0.33/1到0.35/1,以制备Mw是220,000到500,000道尔顿、通常230,000到350,000道尔顿并且更通常250,000到300,000道尔顿的酯化纤维素醚。
如果使用单羧酸的酸酐,那么[脂肪族单羧酸的酸酐/纤维素醚的脱水葡萄糖单元]的摩尔比通常是0.9/1,优选至少1.0/1,并且通常至多8/1,优选至多6/1,并且更优选至多4/1,以制备重量平均分子量Mw是90,000到220,000道尔顿、通常100,000到200,000道尔顿并且更通常100,000到180,000道尔顿的酯化纤维素醚。如果使用单羧酸的酸酐,那么[脂肪族单羧酸的酸酐/纤维素醚的脱水葡萄糖单元]的摩尔比通常是1.2/1到1.4/1,优选1.25/1到1.32/1,以制备Mw是220,000到500,000道尔顿、通常230,000到350,000道尔顿并且更通常250,000到300,000道尔顿的酯化纤维素醚。
本发明方法中所用的纤维素醚的脱水葡萄糖单元的摩尔数可以从用作起始物质的纤维素醚的重量通过由DS(烷氧基)和MS(羟基烷氧基)计算经取代的脱水葡萄糖单元的平均分子量来确定。
纤维素醚的酯化优选在作为反应稀释剂的脂肪族羧酸(如乙酸、丙酸或丁酸)中进行。反应稀释剂可以包含在室温下是液体并且不与纤维素醚反应的少量其它溶剂或稀释剂,如芳香族或脂肪族溶剂,如苯、甲苯、1,4-二噁烷或四氢呋喃;或卤化C1-C3衍生物,如二氯甲烷或二氯甲醚,但脂肪族羧酸的量以反应稀释剂的总重量计优选超过50%、更优选是至少75%并且甚至更优选至少90%。
最优选地,反应稀释剂由脂肪族羧酸组成。因此,下文参考使用脂肪族羧酸作为反应稀释剂来描述酯化方法,但所述方法不限于此。
已发现酯化纤维素醚在20℃下以10wt%丙酮溶液形式测量的粘度可以被酯化反应的三个主要参数影响:1.用作起始物质的纤维素醚的粘度;2.摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元];以及3.摩尔比[酯化催化剂/纤维素醚的脱水葡萄糖单元]。根据本文中的通用传授内容和实例中的更具体传授内容,所属领域的技术人员知晓如何选择酯化反应的这三个主要参数以获得本发明的酯化纤维素醚。
意外地,已发现当将在20℃下以2.0wt%水溶液形式测量粘度为1.20到2.33mPa·s的纤维素醚用作起始物质时,与如现有技术中所公开使用粘度为3mPa·s或大于3mPa·s的纤维素醚时相比,获得以10wt%丙酮溶液测量粘度显著降低的酯化纤维素醚,同时保持其它反应参数恒定。
适当摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元]取决于用作起始物质的纤维素醚的粘度。当用作起始物质的纤维素醚在20℃下以2.0wt%水溶液形式测量的粘度是1.9-2.33mPa·s时,摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元]通常是2.8/1.0或2.8/1.0以上,优选3.0/1.0或3.0/1.0以上,并且更优选3.5/1.0或3.5/1.0以上。当用作起始物质的纤维素醚在20℃下以2.0wt%水溶液测量的粘度仅为1.2-1.8mPa·s时,摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元]可以甚至更低,如0.5/1或0.5/1以上,通常1.0/1或1.0/1以上,更通常1.5/1或1.5/1以上,并且最通常1.7/1或1.7/1以上,同时仍制备在20℃下以10wt%酯化纤维素醚的丙酮溶液形式测量具有上述低粘度的酯化纤维素醚。
如果获得以10wt%丙酮溶液形式测量粘度过高的酯化纤维素醚,那么摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元]应增加,且/或根据本发明的传授内容用作起始物质的纤维素醚在20℃下以2.0wt%水溶液形式测量的粘度应降低。
为获得至少90,000道尔顿、优选至少95,000道尔顿、更优选至少100,000道尔顿并且最优选至少105,000道尔顿的重量平均分子量Mw,视用作起始物质的纤维素醚的粘度而定,摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元]通常是至多8.0/1.0,更通常至多6.0/1.0,甚至更通常至多5.0/1.0,并且最通常至多4.4/1.0。如果其它反应参数保持恒定,那么摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元]越高,酯化纤维素醚的重量平均分子量越低。如果获得具有过低分子量的酯化纤维素醚,那么摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元]应降低,且/或根据本发明的传授内容用作起始物质的纤维素醚在20℃下以2.0wt%水溶液形式测量的粘度应略微增加。然而,选择具有显著较高粘度的纤维素醚作为起始物质将以不可预测方式增加酯化纤维素醚的丙酮粘度。
当酯化纤维素醚的重量平均分子量增加时,其通常意味着其在丙酮中的粘度也增加。但是,已极意外地发现当在20℃下以2.0wt%水溶液形式测量粘度仅为1.20到2.0mPa·s、更优选1.20到1.80mPa·s并且最优选地1.20到1.60mPa·s的纤维素醚用作起始物质时,可以制备具有高重量平均分子量但在20℃下以10wt.%溶液形式测量在丙酮中粘度极低的酯化纤维素醚。
酯化反应通常在酯化催化剂存在下、优选在碱金属羧酸盐(如乙酸钠或乙酸钾)存在下进行。摩尔比[碱金属羧酸盐/纤维素醚的脱水葡萄糖单元]通常是[0.4/1.0]到[3.8/1.0],优选[0.6/1.0]到[2.7/1.0],并且更优选[0.8/1.0]到[1.6/1.0]。如果其它反应参数在指定范围中保持恒定,那么摩尔比[碱金属羧酸盐/纤维素醚的脱水葡萄糖单元]越高,酯化纤维素醚的重量平均分子量一般越高。如果获得以10wt%丙酮溶液形式测量粘度过高的酯化纤维素醚,那么摩尔比[碱金属羧酸盐/纤维素醚的脱水葡萄糖单元]应降低,且/或摩尔比[脂肪族羧酸/纤维素醚的脱水葡萄糖单元]应增加,且/或根据的本发明传授内容用作起始物质的纤维素醚在20℃下以2.0wt%水溶液形式测量的粘度应降低。
通常在60℃到110℃下、优选在70到100℃下加热反应混合物一段时间以足以完成反应,即通常2到25小时,更通常2到8小时。应该充分混合反应混合物以提供均匀反应混合物。完成酯化反应后,反应产物可以从反应混合物以已知方式沉淀,例如通过使其与大量水接触,如美国专利第4,226,981号、国际专利申请案WO 2005/115330或欧洲专利申请案EP 0 219 426中所述。在本发明的优选实施例中,反应产物如2012年3月27日提交的美国临时申请案61/616207或其相应国际专利申请案PCT/US13/030394(以WO 2013/148154公开)中所述由反应混合物沉淀。
本发明的另一方面是包含液体稀释剂和一或多种上述酯化纤维素醚的组合物。如本文所用,术语“液体稀释剂”意指在25℃和大气压下是液体的稀释剂。稀释剂可以是水或有机液体稀释剂或水与有机液体稀释剂的混合物。优选地,液体稀释剂的量足以向组合物提供足够流动性和加工性以用于所需用法(如喷雾干燥)或用于涂布目的。
如本文所用,术语“有机液体稀释剂”意指有机溶剂或两种或两种以上有机溶剂的混合物。优选的有机液体稀释剂是具有一或多个杂原子(如氧、氮或卤素样氯)的极性有机溶剂。更优选的有机液体稀释剂是醇,例如多官能醇,如甘油,或优选单官能醇,如甲醇、乙醇、异丙醇或正丙醇;醚,如四氢呋喃;酮,如丙酮、甲基乙基酮或甲基异丁基酮;乙酸酯,如乙酸乙酯;卤化烃,如二氯甲烷;或腈,如乙腈。
在一个实施例中,本发明的组合物包含单独或与少量水混合的有机稀释剂作为液体稀释剂。在此实施例中,以有机液体稀释剂及水的总重量计,本发明的组合物优选包含超过50、更优选至少65、并且最优选至少75重量%有机液体稀释剂,以及优选少于50、更优选至多35、并且最优选至多25重量%水。本发明的此实施例在本发明包含具有不良水溶性的活性成分时尤其适用。
在另一实施例中,本发明的组合物包含单独或与少量上述有机液体稀释剂混合的水作为液体稀释剂。在此实施例中,以有机液体稀释剂和水的总重量计,本发明的组合物优选包含至少50、更优选至少65、并且最优选至少75重量%水,以及优选至多50、更优选至多35并且最优选至多25重量%有机液体稀释剂。本发明的此实施例尤其适用于由包含本发明的酯化纤维素醚的水性组合物提供包衣或胶囊。在制备水溶液时,式-C(O)-R-COOA的基团的至少一部分优选为其盐形式。
包含液体稀释剂和一或多种上述酯化纤维素醚的本发明的组合物适用作活性成分的赋形剂系统,并且尤其适用作制备活性成分的赋形剂系统的中间物,其中活性成分如肥料、除草剂或杀虫剂,或生物活性成分,如维生素、草药和矿物补充剂以及药物。因此,本发明的组合物优选包含一或多种活性成分,最优选一或多种药物。术语“药物”是常规的,其表示在投与动物、尤其人类时具有有益的预防和/或治疗特性的化合物。优选地,药物是「低溶解性药物」,意指药物在生理学相关pH值(例如pH 1-8)下的水溶性是约0.5mg/mL或0.5mg/mL以下。本发明在药物的水溶性降低时较为适用。因此,本发明的组合物优选用于水溶性小于0.1mg/mL或小于0.05mg/mL或小于0.02mg/mL或甚至小于0.01mg/mL的低溶解性药物,其中水溶性(mg/mL)是在任何生理学相关水溶液(例如pH值是1到8的生理学相关水溶液,包括USP模拟胃和肠缓冲液)中观察到的最小值。为了得益于本发明,活性成分不一定是低溶解性活性成分,但低溶解性活性成分代表适用于本发明的优选类别。在所要使用环境中展现可观水溶性的活性成分的水溶性可能高达1到2mg/mL,或甚至高达20到40mg/mL。适用的低溶解性药物列于国际专利申请案WO 2005/115330,第17-22页中。
本发明的液体组合物以组合物的总重量计优选包含1到40%、更优选5到35%、甚至更优选7到30%并且最优选10到25%至少一种上述酯化纤维素醚;40到99%、更优选50到94.9%、甚至更优选65到92.5%并且最优选70到89%上文另外描述的液体稀释剂;以及0到40%、更优选0.1到40%、甚至更优选0.5到25%并且最优选1到15%活性成分。酯化纤维素醚在20℃下以10wt%丙酮溶液形式测量的低粘度使得可以并入高浓度的酯化纤维素醚,即,高的酯化纤维素醚与液体稀释剂比率,同时仍提供具有适当低粘度的液体组合物。这可以以两种方式使用来制备活性成分在酯化纤维素醚中的固体分散体:1.酯化纤维素醚/活性成分的比率保持与在已知的较稀组合物中相同。在此情形下,较高浓度的酯化纤维素醚也使液体组合物中活性成分的浓度较高,并且,因此在制备固体分散体中使活性成分的产量增加,同时维持活性成分的相同稳定性。2.或者,仅液体组合物中酯化纤维素醚的浓度提高,但活性成分的浓度不提高。这得到较高的酯化纤维素醚/活性成分比率,从而使移除液体稀释剂后酯化纤维素醚的基质中的活性成分的稳定改良而不会降低活性成分的产量。这意味着配制者可以在液体配制物中较高含量的酯化纤维素醚下操作-而无需降低活性成分的含量-以使活性成分在固体剂型中的非晶形态的稳定增强。本发明的酯化纤维素醚允许液体组合物中活性成分的负载较高,同时在制备固体分散体中仍获得适当高产量。如WO2004/014342,第13页,最后一段中所提议,不必制备半有序分散液替代非晶形分散液来获得较高活性成分产量。
在本发明的一个方面中,包含至少一种上述酯化纤维素醚、一或多种活性成分和任选地一或多种佐剂的组合物可以液体形式、例如悬浮液、浆液、可喷雾组合物或糖浆形式使用。液体组合物适用于例如经口、经眼、局部、经直肠或经鼻施用。液体稀释剂通常应该是药物学上可接受的,如乙醇或甘油,任选地如上所述与水混合。酯化纤维素醚在丙酮或另一种有机溶剂中的低粘度显著改良液体组合物的处理,如其被倾倒或泵送的能力。
在本发明的另一个方面,本发明的液体组合物用于制备固体分散体,其包含至少一种活性成分(如上文另外描述的药物)、至少一种上述酯化纤维素醚以及任选地一或多种佐剂。固体分散体通过从组合物移除液体稀释剂产生。酯化纤维素醚在丙酮或另一种有机溶剂中的低粘度使得可向组合物中并入高浓度的酯化纤维素醚和因此高浓度的药物,同时仍维持液体组合物的适当低粘度。这在液体组合物用于涂布目的时或在包含酯化纤维素醚的液体组合物经受喷雾干燥(例如用于制备包含活性成分和酯化纤维素醚的固体分散体)时高度有利于获得高产量。此外,可以通过喷雾干燥配制上述使用高酯化纤维素醚与活性成分比率的液体配制物。需要高酯化纤维素醚与活性成分比率来维持难溶性活性成分的过饱和中和用于提高其生物可用性。
一种从液体组合物移除液体稀释剂的方法为通过将所述液体组合物浇铸成膜或胶囊或通过将液体组合物涂覆到又可以包含活性成分的固体载体上。本发明的液体组合物用于涂布目的的用途是本发明的优选方面。
制备固体分散体的优选方法是通过喷雾干燥。术语“喷雾干燥”指涉及使液体混合物破碎成小液滴(雾化),并且在存在自液滴蒸发溶剂的强驱动力的喷雾干燥装置中从混合物快速移除溶剂的方法。喷雾干燥方法和喷雾干燥设备通常描述在佩里的《化学工程手册》,第20-54页到第20-57页(第六版,1984年)(Perry′s Chemical Engineers′Handbook,pages 20-54to 20-57(Sixth Edition 1984))中。关于喷雾干燥方法和设备的更多细节由马歇尔,“雾化和喷雾干燥”,50,《化学工程进展专著系列2》,(1954)(Marshall,″Atomization and Spray-Drying,″50Chem.Eng.Prog.Monogr.Series 2(1954))和马斯特,《喷雾干燥手册》(第四版,1985)(Masters,Spray Drying Handbook(Fourth Edition 1985))综述。适用的喷雾干燥方法描述在国际专利申请案WO 2005/115330,第34页,第7行-第35页,第25行中。或者,本发明的固体分散体可以通过如下步骤制备:i)掺合a)至少一种如上文所定义的酯化纤维素醚、b)一或多种活性成分和c)一或多种任选的添加剂,以及ii)使掺合物经受挤压。如本文所用,术语“挤压”包括称为射出模制、熔融浇铸和压缩模制的方法。挤压、优选熔融挤压包含活性成分(如药物)的组合物的技术是已知的,并且由约尔格·布赖滕巴赫,熔融挤压:从方法到药物传递技术,《欧洲药剂学和生物药剂学杂志》,54(2002)107-117(Joerg Breitenbach,Melt extrusion:from process to drug delivery technology,European Journal of Pharmaceutics and Biopharmaceutics 54(2002)107-117)描述或描述在欧洲专利申请案EP 0 872 233中。本发明的固体分散体以酯化纤维素醚a)和活性成分b)的总重量计优选包含20到99.9%、更优选30到98%并且最优选60到95%上述酯化纤维素醚a),以及优选0.1到80%、更优选2到70%并且最优选5到40%活性成分b)。以固体分散体的总重量计,酯化纤维素醚a)与活性成分b)的组合量优选是至少70%,更优选至少80%并且最优选至少90%。剩余量(如果存在的话)由一或多种下述佐剂c)组成。固体分散体可以包含一或多种酯化纤维素醚a)、一或多种活性成分b)以及任选地一或多种佐剂c),但是其总量通常在上述范围内。
在至少一种酯化纤维素醚中包含至少一种活性成分的固体分散体形成后,可以使用若干加工操作来促进将分散液并入剂型中。这些处理操作包括干燥、造粒以及研磨。在固体分散体中包括任选的佐剂可能适用于将组合物配制成剂型。本发明的固体分散体可以是各种形式,如丝束、丸粒、颗粒、丸剂、片剂、囊片、微粒、胶囊或射出模制胶囊的填充物,或粉末、膜、糊剂、乳膏、悬浮液或浆液形式。
以剂型的总重量计,剂型中活性成分的量通常是至少0.1%、优选至少1%、更优选至少3%、最优选至少5%并且通常至多70%、优选至多50%、更优选至多30%、最优选至多25%。
在本发明的另一个方面,包含液体稀释剂和一或多种上述酯化纤维素醚的本发明的组合物可以用于涂布剂型,如片剂、颗粒、丸粒、囊片、糖锭、栓剂、阴道栓剂或可植入剂型以形成经涂布组合物。如果本发明的组合物包含活性成分(如药物),那么可以实现药物分层,即,剂型和涂层可以包含用于不同最终用途和/或具有不同释放动力学的不同活性成分。
在本发明的另一个方面,包含液体稀释剂和一或多种上述酯化纤维素醚的本发明的组合物可以用于在包含使液体组合物与浸渍销接触的步骤的方法中制造胶囊。
本发明的液体组合物和固体分散体可以另外包含任选的添加剂,如着色剂、颜料、遮光剂、风味和口味改良剂、抗氧化剂以及其任何组合。任选的添加剂优选是药物学上可接受的。一或多种任选的佐剂的适用量和类型在所属领域中通常是已知的,并且取决于本发明的液体组合物或固体分散体的既定最终用途。
本发明的一些实施例现将详细地描述于以下实例中。
实例
除非另外提到,否则所有份数和百分比都以重量计。在实例中,使用以下测试程序。
羟基丙基甲基纤维素(HPMC)样品的粘度
HPMC样品的粘度在20℃±0.1℃下以2.0重量%水溶液形式测量。2.0重量%HPMC水溶液根据美国药典(United States Pharmacopeia)(USP 35,“羟丙甲纤维素(Hypromellose)”,第3467-3469页)制备,接着根据DIN 51562-1:1999-01(1999年1月)进行乌氏粘度测量。
羟基丙基甲基纤维素乙酸酯丁二酸酯(HPMCAS)的粘度
2.0重量%HPMC在0.43wt%NaOH水溶液中的溶液如“羟丙甲纤维素乙酸酯丁二酸酯”,《美国药典和国家处方集》,NF 29,第1548-1550页所述制备,接着在20℃下根据DIN 51562-1:1999-01(1999年1月)进行乌氏粘度测量。
10wt%酯化纤维素醚在丙酮中的溶液通过如下步骤制备,首先根据“羟丙甲纤维素乙酸酯丁二酸酯”,《美国药典和国家处方集》,NF 29,第1548-1550页测定HPMCAS的干燥失重。随后在室温下将以干燥重量计10.00g HPMCAS与100g丙酮在剧烈搅拌下混合。将混合物在辊式混合器上辊压约24小时。使用米佳夫(Megafuge)1.0离心机(可购自德国贺利氏控股公司(Heraeus Holding GmbH,Germany))在2000rpm下离心溶液3分钟,接着在20℃下根据DIN 51562-1:1999-01(1999年1月)进行乌氏粘度测量。
HPMCAS的醚基和酯基的含量
酯化纤维素醚中醚基的含量以如对于“羟丙甲纤维素”,《美国药典和国家处方集》,USP 35,第3467-3469页(“Hypromellose”,United States Pharmacopeia and National Formulary,USP 35,pp 3467-3469)所述相同的方式确定。
根据羟丙甲纤维素乙酸酯丁二酸酯,《美国药典和国家处方集》,NF 29,第1548-1550页(Hypromellose Acetate Succinate,United States Pharmacopeia and National Formulary,NF 29,pp.1548-1550)确定用乙酰基(-CO-CH3)进行的酯取代和用丁二酰基(-CO-CH2-CH2-COOH)进行的酯取代。将酯取代的报告值针对挥发物进行校正(如在上述HPMCAS专论中的章节“干燥失重”中所述确定)。
HPMCAS的Mw、Mn和Mz的测定
除非另外说明,否则Mw、Mn和Mz是根据《医药和生物医学分析杂志》,56(2011)743(Pharmaceutical and Biomedical Analysis 56(2011)743)测量流动相是40体积份乙腈与60体积份含有50mM NaH2PO4和0.1M NaNO3的水性缓冲液的混合物。将流动相调节到pH 8.0。经由0.45μm孔径的针筒过滤器将纤维素醚酯溶液过滤到HPLC小瓶中。
更确切地说,所用化学品和溶剂是:
聚氧化乙烯标准材料(简称为PEOX 20 K和PEOX 30 K)购自加利福尼亚州帕洛阿尔托市安捷伦技术公司(Agilent Technologies,Inc.Palo Alto,CA),目录号PL2083-1005和PL2083-2005。
乙腈(HPLC级≥99.9%,曲马索普拉斯(CHROMASOL plus)),目录号34998;氢氧化钠(半导体级,99.99%,以痕量金属计),目录号306576;水(HPLC级,曲马索乌普拉斯(CHROMASOLV Plus))目录号34877;以及硝酸钠(99,995%,以痕量金属计),目录号229938购自瑞士西格玛-奥德里奇公司(Sigma-Aldrich,Switzerland)。
磷酸二氢钠(≥99.999%,崔斯力特(TraceSelect))目录号71492购自瑞士福鲁卡公司(FLUKA,Switzerland)。
PEOX20 K的5mg/mL标准化溶液、PEOX30 K的2mg/mL标准溶液和HPMCAS的2mg/mL样品溶液通过向小瓶中添加称量量的聚合物并将其用测量体积的流动相溶解来制备。在室温下在使用涂布有PTFE的磁力搅拌棒搅拌下使所有溶液在加盖小瓶中溶解24小时。
经由0.02μm孔径和25mm直径的针筒过滤器(沃特曼公司(Whatman)阿拉特(Anatop)25,目录号6809-2002)沃特曼(Whatman)将标准化溶液(PEOX 20k,单一制剂,N)和标准溶液(PEOX30 K,双制剂,S1和S2)过滤到HPLC小瓶中。
将测试样品溶液(HPMCAS,制备两份,T1、T2)和实验室标准品(HPMCAS,单一制剂,LS)经由0.45μm孔径的针筒过滤器(尼龙,例如艾克迪克(Acrodisc)13mm VWR目录号514-4010)过滤到HPLC小瓶中。
色谱条件和操作工序如陈,R.等人;《医药和生物医学分析杂志》,56(2011)743-748(Chen,R.et al.;Journal of Pharmaceutical and Biomedical Analysis 56(2011)743-748)所描述进行。SEC MALLS仪器装备包括HP1100系统(来自加利福尼亚州帕洛阿尔托市安捷伦技术公司(Agilent Technologies,Inc.Palo Alto,CA));道恩公司(DAWN)海莱士(Heleos)II 18个角度激光散射检测器和奥普提拉公司(OPTILAB)雷克斯(rex)折射率检测器,两者都来自加利福尼亚州圣巴巴拉市怀亚特技术公司(Wyatt Technologies,Inc.Santa Barbara,CA)。分析型尺寸排阻柱( GMPWXL,300×7.8mm)购自东曹生物科学公司(Tosoh Bioscience)。奥普提拉公司和道恩公司的产品都在35℃下操作。分析型SEC柱在室温(24±5℃)下操作。流动相是40体积份乙腈和60体积份含有50mM NaH2PO4和0.1M NaNO3的水性缓冲液的混合物,其如下制备:
水性缓冲液:在洁净的2L玻璃瓶中,在搅拌下将7.20g磷酸二氢钠和10.2g硝酸钠添加到1.2L纯化水中直到溶解为止。
流动相:将800mL乙腈添加到1.2L如上制备的水性缓冲液中,并搅拌直到获得良好混合物并且温度平衡到环境温度为止。
用10M NaOH将流动相的pH值调节到8.0,并经由0.2m尼龙膜过滤器过滤。流速是0.5mL/min,并进行在线脱气。注射体积是100μL并且分析时间是35min。
收集MALLS数据,并使用对于HPMCAS而言0.120ml/g的dn/dc值(折射率增量)用怀亚特公司阿斯特拉软件(版本5.3.4.20)处理。在分子量计算中不使用检测器第1-4、17和18号的光散射信号。代表性色谱操作工序如下所给:B、N、LS、S1(5x)、S2、T1(2x)、T2(2x)、T3(2x)、T4(2x)、S2、T5(2x)等,S2、LS、W,其中B表示空白注射的流动相,N1表示标准化溶液;LS表示实验室标准品HPMCAS;S1和S2分别表示标准溶液1和2;T1、T2、T3、T4和T5表示测试样品溶液,并且W表示注射水。(2x)和(5x)表示注射相同溶液的次数。
奥普提拉公司和道恩公司的产品都根据制造商的推荐程序和频率定期校准。对于每个操作工序,注射100μL 5mg/mL聚氧化乙烯标准品(PEOX20K)来用于相对于90°检测器标准化所有角度光散射检测器。
使用此单分散聚合物标准品也使得可以确定奥普提拉公司产品与道恩公司产品之间的体积延迟,从而使得可以将光散射信号与折射率信号进行正确对准。这对于计算每个数据片的重量平均分子量(Mw)是必需的。
制备实例1-4的羟基丙基甲基纤维素乙酸酯丁二酸酯(HPMCAS)
在充分搅拌下将下表1中所列量的冰醋酸、乙酸酐、羟基丙基甲基纤维素(HPMC)、丁二酸酐和乙酸钠(无水)引入反应容器中,得到均匀反应混合物。HPMC具有下表2中所列的甲氧基和羟基丙氧基取代度和在20℃下以2%水溶液形式测量的粘度。
如下表1中所列,在85℃下在搅拌下加热混合物3或3.5小时以实现酯化。在搅拌下将x L水添加到反应器中以使HPMCAS沉淀。从反应器移出沉淀的产物,并用yL水通过使用在5200rpm下操作的超特拉斯(Ultra-Turrax)搅拌棒S50-G45施加高剪切混合来洗涤。洗涤分若干部分进行,其中具有中间过滤步骤以获得具有极高纯度的HPMCAS。应该洗涤HPMCAS产物并过滤直到其粘度基本上恒定为止(10wt.%丙酮溶液)。水的升数x和y列于下表1中。最终过滤步骤后,在50℃下干燥产物隔夜。
重复实例4。得到基本上相同结果的两个实验的平均值列于下表2中。
制备比较实例A和B的HPMCAS
根据比较实例A和B的HPMCAS的制备如实例1到4中进行,但使用如欧洲专利申请案EP 0219 426 A2的实例2中所公开的HPMC类型和冰醋酸、乙酸酐、HPMC、丁二酸酐和乙酸钠(无水)的重量比。使用量列于下表1中。
比较实例A中所用的HPMC在20℃下以2%水溶液形式测量的粘度是6.0mPa·s,所述HPMC具有28.2重量%羟基丙氧基和9.0重量%甲氧基。此HPMC可以美多秀E6 LV优质纤维素醚购自陶氏化学公司。比较实例B中所用的HPMC在20℃下以2%水溶液形式测量的粘度是3.1mPa·s,所述HPMC具有9.3重量%羟基丙氧基和28.2重量%甲氧基。此HPMC可以美多秀E3 LV优质纤维素醚购自陶氏化学公司。
在85℃下在搅拌下加热混合物3.5小时以实现酯化。在搅拌下将xL水添加到反应器中以使HPMCAS沉淀。从反应器移出沉淀的产物,并用yL水通过使用在5200rpm下操作的超特拉斯搅拌棒S50-G45施加高剪切混合来洗涤。水的数量x和y列于下表1中。产物通过过滤分离并在55℃下干燥12小时。
比较实例A和B中的所得酯取代度乙酰基%和丁二酰基%明显不同于欧洲专利申请案EP 0219 426 A2的实例2中所公开的结果。因此,重复比较实例A和B。在重复的实例A和B中所得酯取代度乙酰基%和丁二酰基%基本上与第一组比较实例A和B中相同。表2中的结果展示比较实例A和B的两次操作的平均值。
制备比较实例C和D的HPMCAS
根据比较实例C和D的HPMCAS的制备如实例1到4中进行,但使用如国际专利申请案WO 2005/115330,第51和52页,聚合物1和3所公开的冰醋酸、乙酸酐、HPMC、丁二酸酐和乙酸钠(无水)的重量比。产物如国际专利申请案WO 2005/115330中所述获得、分离并洗涤。使反应混合物在2.4L水中淬灭,从而使聚合物沉淀。再使用1L水完成沉淀,例如仅沉淀C。随后分离聚合物并用3×300mL水洗涤。接着将聚合物溶解在600mL丙酮中并再次在2.4L水中沉淀。为了完成沉淀,再添加1L水。
比较实例E到G
如国际专利申请案WO 2011/159626第1页和第2页中所公开,HPMCAS目前可购自信越化学公司(Shin-Etsu Chemical Co,Ltd.)(日本东京(Tokyo,Japan)),其商标名为“AQOAT”。信越公司制造了具有不同取代基含量组合的三种等级的AQOAT聚合物以在各种pH值下提供肠保护:AS-L、AS-M和AS-H,其通常后面跟着标识“F”(用于表示精细)或“G”,如AS-LF或AS-LG。其销售说明如下所列。
WO 2011/159626中所列的AQOAT聚合物的特性
市售材料的样品如上文进一步描述来分析。
比较实例H、I-1、I-2、J-1和J-2的HPMCAS
HPMCAS样品如WO 2011/159626的第34页和第35页所述制备。在比较实例H中,HPMCAS-K(1)的配方完全重复。在比较实例I-1和I-2中,HPMCAS-K(2)的配方和在比较实例J-1和J-2中,HPMCAS-K(3)的配方完全重复。比较实例I和J各自进行两次,并分别报告为I-1、I-2、J-1和J-2,因为比较实例I-1和I-1中关于DOSAc和DOSs的结果与WO 2011/159626中关于HPMCAS-K(2)和HPMCAS-K(3)报告的结果有偏差。
根据实例1-4和比较实例A-D、H-、I-1、I-2、J-1和J-2制备的HPMCAS的特性和市售比较实例E到G的特性列于下表2中。
在下表2中,缩写具有以下含义:
DSM=DS(甲氧基):甲氧基的取代度;
MSHP=MS(羟基丙氧基):羟基丙氧基的摩尔取代度;
DOSAc:乙酰基的取代度;
DOSs:丁二酰基的取代度
表1
*以干燥形式计算
表2
*粘度在20℃下以2.0wt%酯化纤维素醚在0.43wt%NaOH水溶液中的溶液形式测量
**两个各别制备实验的平均值
欧洲专利申请案EP 0 219 426公开比较实例A的HPMCAS适用作片剂上的肠溶薄膜衣材料,其可抵抗拟胃液,而涂布有比较实例B的HPMCAS的片剂在拟胃液中崩解。比较实例A的HPMCAS的重量平均分子量高于比较实例B。显然极其需要高重量平均分子量的HPMCAS,但比较实例A的HPMCAS以10wt.%丙酮溶液形式测量的粘度高得多,并且在喷雾干燥或涂布方法中处理起来不太有效。
一方面实例1-4与另一方面比较实例A和E-G之间的比较说明实例1-4的HPMCAS的重量平均分子量在与比较实例E-G的HPMCAS相同的范围内或甚至高于比较实例E-G的HPMCAS,但以10wt.%丙酮溶液形式测量的粘度低得多。
一方面实例1-3与另一方面比较实例B-D之间的比较说明实例1-4的HPMCAS的重量平均分子量高于比较实例B-D的HPMCAS,并且实例1-4的HPMCAS展现的10%丙酮溶液粘度与比较实例B-D的10%丙酮溶液粘度相当或甚至明显低于所述粘度。
在10wt-%浓度下,比较实例N、O-1、O-2、P-1和P-2的HPMCAS不溶或仅部分可溶于丙酮中。在比较实例I-1和I-2中,所用HPLC方法中的回收率过低而不能进行相当可靠地Mw、Mn和Mz测定。回收率=[从HPLC柱回收的HPMCAS的重量/引入HPLC柱中的HPMCAS的重量]×100。
具有低粘度和高分子量的新颖酯化纤维素醚专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




















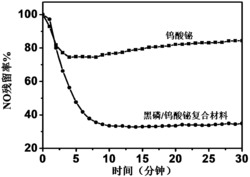
动态评分
0.0