









专利摘要
本发明公开了一种无机钙钛矿量子点的固相合成方法,即通过研磨含Cs、Pb、卤素(Cl,Br,I)的前驱玻璃,促使其发生力致晶化。该方法克服了传统液相法面临的成本、环境友好性、规模化制备等诸多难题。而非晶玻璃基体对CsPbX3量子点的包裹可大幅提高材料的热/化学稳定性。
权利要求
1.一种无机钙钛矿量子点的固相合成方法,其特征在于,包括以下步骤:
1)按照一定比例将含铅化合物、含卤化合物、含铯化合物、以及玻璃基体组合物混合、熔融,随后进行熔体急冷,得到前驱玻璃;
2)将步骤1)得到的前驱玻璃块体进行破碎,而后进一步研磨,使之发生玻璃力致晶化,获得镶嵌CsPbX
3)作为选择,可将步骤2)得到的微晶玻璃粉进一步置于玻璃转变点温度之上进行一定时间的热处理,以提高CsPbX
2.根据权利要求1所述的无机钙钛矿量子点的固相合成方法,其特征在于:步骤1)中,含卤化合物中的卤族元素为Cl,Br,I中任一种或任两种;玻璃基体组合物包括但不限于SiO
3.根据权利要求1所述的无机钙钛矿量子点的固相合成方法,其特征在于:在步骤1)中,将各组分的粉体原料混合后,在玛瑙球磨罐中混合并研磨,研磨均匀后置于坩埚中,加热使之熔融,得到玻璃熔体。
4.根据权利要求1所述的无机钙钛矿量子点的固相合成方法,其特征在于:玻璃粉体原料加热到500~800℃,保温0.1~2小时,得到玻璃熔体。
5.根据权利要求4所述的无机钙钛矿量子点的固相合成方法,其特征在于:玻璃粉体原料加热到600~700℃,保温0.5~1小时,效果为佳。
6.根据权利要求1所述的无机钙钛矿量子点的固相合成方法,其特征在于:将玻璃熔体快速倒入模具中成型,得到块状前驱玻璃。
7.根据权利要求6所述的无机钙钛矿量子点的固相合成方法,其特征在于:可将所得的前驱玻璃进一步放入电阻炉中退火,以消除内应力。
8.根据权利要求1所述的无机钙钛矿量子点的固相合成方法,其特征在于:在步骤2)中,将步骤1)得到的前驱玻璃块体置于玻璃破碎机中碾碎成小颗粒,进而将这些玻璃颗粒放入球磨机中进一步研细成微米级的微晶玻璃粉。
9.根据权利要求8所述的无机钙钛矿量子点的固相合成方法,其特征在于:球磨机转数为100-600转/分钟,球磨时间为10分钟-10小时。
10.根据权利要求9所述的无机钙钛矿量子点的固相合成方法,其特征在于:球磨机为400-500转/分钟,球磨时间为为1-3小时,效果为佳。
11.根据权利要求1所述的无机钙钛矿量子点的固相合成方法,其特征在于:在步骤3)中,可将步骤2)得到的微晶玻璃粉放入电阻炉中加热至300~600℃,保温0.5~12小时,以使玻璃基体内部发生进一步晶化,提高CsPbX
12.根据权利要求11所述的无机钙钛矿量子点的固相合成方法,其特征在于:微晶玻璃粉放入电阻炉中加热至400~500℃,保温2~6小时,效果为佳。
13.一种根据权利要求1-12中任一项所述的固相合成方法所制备的钙钛矿量子点材料,其结构特征在于:CsPbX
14.一种根据权利要求1-12中任一项所述的固相合成方法所制备的钙钛矿量子点材料,用于背光显示和应力传感领域。
说明书
技术领域
本发明涉及固体发光材料制备技术领域,尤其是涉及一种无机钙钛矿量子点的固相合成方法。
背景技术
半导体量子点是应用于背光显示领域的一类重要光转换材料。传统的基于硫属化合物的半导体量子点,制备过程繁琐(需构建核壳结构),含有有毒的Cd元素,而且由于半峰宽较宽,色纯度低,不利于拓宽背光光源的色域。近年来,一类具有钙钛矿结构的离子性半导体量子点CsPbX3(X为Cl,Br,I中任一种)引起人们广泛的关注,其制备方法相对简单,特别是,荧光量子效率高(可达90%以上),半峰宽窄(20-30纳米),重吸收效应小,颜色可调,已然成为当下背光领域的明星材料。2015年,L.Protesescu等人率先报道了采用高温热注入法制备钙钛矿量子点(Nano Lett.,2015,15,3692)。2016年,X.M.Li等人基于过饱和析晶法实现了钙钛矿量子点快捷制备(Adv.Funct.Mater.,2016,26,2435)。2016年,T.Yu等人采用微波法合成了钙钛矿量子点(Angew.Chem.Int.Ed.2016,55,1)。必须指出地是,上述合成方法均为液相法,需使用到大量对环境有害的有机试剂,合成成本昂贵,且无法实现批量化制备;另一方面,由于钙钛矿量子点的热/化学稳定性较差,其实际应用受限。
针对现有技术存在的问题,本发明提供了一种钙钛矿量子点的新型固相合成技术路线。首先,将含铅化合物、含卤化合物(卤素为Cl,Br,I中任一种或任两种)、含铯化合物,与常见玻璃基体组合物(如SiO2,Al2O3,P2O5,B2O3,TeO2,GeO2,ZnO,Na2O,K2O,Li2O,CaO,SrO,BaO等)以一定比例进行共混,置于坩埚中,并放入电阻炉中进行烧制,将熔体快速冷却后形成透明玻璃。随后,将获得的玻璃块体敲碎、研磨,形成微米级粉末。由于发生玻璃的力致晶化现象,CsPbX3(X为Cl,Br,I中任一种或任两种)量子点在玻璃基体中析出,因而获得的粉末为镶嵌CsPbX3量子点的微晶玻璃粉。为了进一步提高CsPbX3量子点在玻璃基体中的结晶含量,可进一步将上述微晶玻璃粉置于玻璃转变点温度之上进行一定时间的热处理。值得一提的是,基于该固相法合成技术获得钙钛矿量子点材料,制备方法简便,绿色环保,不产生废水废液,成本低廉,可实现规模化制备;而且受益于非晶玻璃基体的保护,CsPbX3量子点的热/化学稳定性大幅度提高。
发明内容
本发明提供了一种无机钙钛矿量子点的固相合成方法。本发明的无机钙钛矿量子点材料为镶嵌CsPbX3量子点的微晶玻璃,其中:
X为Cl,Br,I中任一种或任两种。
所述镶嵌CsPbX3量子点的微晶玻璃,其玻璃基体由包含下列组分的化合物制备:
含铅化合物、含卤化合物(卤素为Cl,Br,I中任一种或任两种)、含铯化合物、以及玻璃基体组合物(如SiO2,Al2O3,P2O5,B2O3,TeO2,GeO2,ZnO,Na2O,K2O,Li2O,CaO,SrO,BaO等)
作为实例,本发明镶嵌CsPbX3量子点的微晶玻璃可以包含摩尔比如下的各组分:
45P2O5-20PbBr2-10NaBr-13Cs2O-12SrO;或
50P2O5-10PbBr2-5PbI2-10NaBr-10Cs2O-15CaO;或
30B2O3-30SiO2-10ZnO-5BaO-5Cs2O-5PbBr2-5PbI2-5NaI–5NaBr;或
50GeO2-10B2O3-5ZnO-5BaO-5K2O-5Cs2O-10PbCl2-10NaCl;或
50TeO2-5Al2O3-10B2O3-5ZnO-5Na2O-5BaO-5Cs2O-10PbBr2-5NaBr;或
30P2O5-25SiO2-15ZnO-5BaO-5Cs2O-10PbI2-10NaI;或
…
本发明还提供了上述镶嵌CsPbX3量子点微晶玻璃的制备方法,包括以下步骤:
1)按照一定比例将各种化合物混合、熔融,随后进行熔体急冷,得到前驱玻璃;
2)将步骤1)得到的前驱玻璃块体进行破碎,而后进一步研磨,过筛。由于发生玻璃力致晶化现象,获得的粉末为镶嵌CsPbX3量子点的微晶玻璃粉。
3)可将步骤2)得到的微晶玻璃粉进一步置于玻璃转变点温度之上进行一定时间的热处理,以提高CsPbX3量子点在玻璃基体中的结晶含量。
根据本发明的制备方法,其步骤1)中:
将各组分的粉体原料混合后,例如在玛瑙球磨罐中混合并研磨,研磨均匀后置于坩埚中,加热使之熔融,得到玻璃熔体;
根据本发明,优选地,在电阻炉中加热到500~800℃,优选600-700℃,保温0.1-2小时,优选0.5-1小时,以使粉体原料熔融,得到玻璃熔体;
作为实例,可将玻璃熔体快速倒入模具中成型,得到块状前驱玻璃;或者,作为选择,可将所得的前驱玻璃退火,例如放入电阻炉中退火,以消除内应力;例如,退火的温度可以为100~300℃;进一步优选为150~250℃。
步骤2)中:
将步骤1)得到的前驱玻璃块体置于玻璃破碎机中碾碎成小颗粒,进而将这些玻璃颗粒放入球磨机中进一步研细成微米级的微晶玻璃粉。球磨机转数为100-600转/分钟,优选为400-500转/分钟;球磨时间为10分钟-10小时,优选为1-3小时。
步骤3)中:
将步骤2)得到的微晶玻璃粉放入电阻炉中加热至300~600℃,例如400~500℃;优选地,可在加热后保温0.5~12小时,优选2~6小时,以使玻璃基体内部发生进一步晶化,提高CsPbX3量子点的结晶含量。
本发明玻璃的力致晶化现象需在特定玻璃组分下方可观察到。玻璃组分不同,其相应的玻璃网络结构、分相结构、和晶化势垒均不同,从而将强烈影响CsPbX3量子点的析晶行为。实验表明,倘若玻璃网络结构过于紧密、晶化势垒过高、或者无分相结构,都无法观察到玻璃力致晶化现象。
本发明玻璃的力致晶化现象是基于所述玻璃疏松的网络结构、玻璃分相,以及CsPbX3量子点离子特性的综合效应所引起的。机械力作用于玻璃使其发生断裂或摩擦,将破坏玻璃网络结构,而网络中化学键断裂释放的能量可促使CsPbX3量子点的成核和生长;同时,断裂剪切力使得玻璃剪切带之间产生相对滑移,增加了玻璃组元相互接触的几率,从而降低了组元迁移的晶化势垒。此外,前驱玻璃中弥散分布着液滴状Cs、Pb、和Br元素的富集区,这种玻璃分相结构的存在,使得玻璃晶化更易发生。玻璃的力致晶化现象主要发生在玻璃表面。
本发明经研磨获得微晶玻璃粉继续进行晶化热处理,可提高CsPbX3量子点在玻璃基体中的结晶含量。其晶化机制为,加热玻璃基体至玻璃化转变温度点之上,玻璃性质将发生剧烈变化——玻璃网络结构发生弛豫,玻璃粘度下降,这将有助于玻璃组元发生迁移。当提供的热能高于玻璃晶化激活能时,玻璃基体内部将产生CsPbX3量子点的结晶。
根据本发明的制备方法,所使用的坩埚可以是石英坩埚、石墨坩埚、铂金坩埚或者刚玉坩埚。
本发明还提供所述玻璃用作荧光材料的用途。特别地,本发明所述的微晶玻璃可应用于背光材料以及应力传感。
本发明的有益效果:
本发明创造性地提出了一种无机钙钛矿量子点的固相合成方法,所获材料的结构特征是CsPbX3量子点弥散分布于无机非晶玻璃基体之中。全固相合成克服了传统液相合成面临的成本、环境友好性、规模化制备等诸多难题。而非晶玻璃基体对CsPbX3量子点的包裹可大幅提高其热/化学稳定性。
附图说明
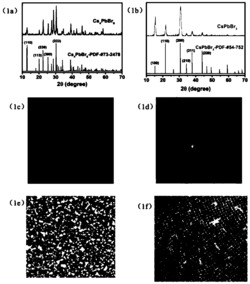
图1是实施例1中CsPbBr3微晶玻璃粉末的X射线衍射图;
图2是实施例1中CsPbBr3微晶玻璃粉末的扫描电镜图;
图3是实施例1中CsPbBr3微晶玻璃粉末上单个颗粒的元素分析;
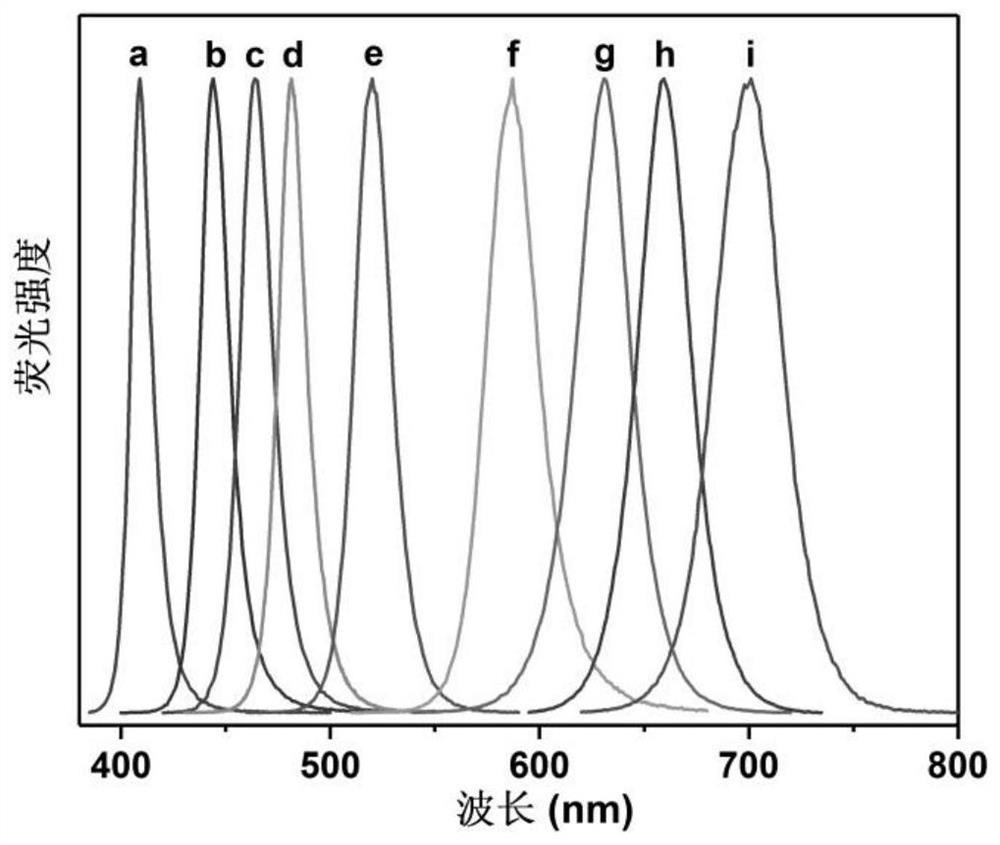
图4是实施例1中CsPbBr3-xIx固溶体量子点微晶玻璃粉的室温发射谱;
图5是实施例1中CsPbBr3微晶玻璃粉末在热处理前后的X射线衍射图;
图6是实施例1中CsPbBr3微晶玻璃粉末在热处理前后的室温发射谱;
具体实施方式
以下通过示例性的具体实施例对本发明的技术方案进行详细说明。但不应将这些实施例解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
除非另有说明,实施例中所记载的原料及试剂均为市售产品。
实施例1
将分析纯的P2O5,PbBr2,NaBr,Cs2O,SrO粉体,按45P2O5:20PbBr2:10NaBr:13Cs2O:12SrO(摩尔百分比)的配比精确称量后置于研钵中,混合并研磨均匀后置于氧化铝坩埚中,放入电阻炉中加热到600℃后保温2小时使之熔融,而后,将玻璃熔体取出并快速倒入模具中成形,得到块状前驱玻璃,最后,将获得的前驱玻璃放入电阻炉中在200℃退火以消除内应力。将得到的前驱玻璃块体置于玻璃破碎机中碾碎成小颗粒,进而将这些玻璃颗粒放入球磨机中进一步研细成微米级的微晶玻璃粉。球磨机转数为480转/分钟;球磨时间为2小时。
图1中X射线衍射数据(扫描范围θ:10-90度)表明,所获微晶玻璃粉末中镶嵌CsPbBr3晶相。
扫描电镜结果表明CsPbBr3纳米晶均匀分布在玻璃基体中(如图2所示)。针对玻璃表面上单颗晶粒进行元素分析,可确认该晶粒的组成含Cs,Pb,Br三种元素(如图3所示)。通过改变PbCl2/PbBr2/PbI2的投料比,可分别获得镶嵌CsPbBr3-xIx、CsPbBr3-xClx、和CsPbCl3-xIx(x=0-3)固溶体量子点的微晶玻璃粉。作为示例,用FLS920荧光光谱仪测量CsPbBr3-xIx固溶体量子点微晶玻璃粉的室温发射谱(如图4所示)。在460纳米激发的发射谱上,出现了典型的钙钛矿量子点的窄带激子发射峰,其发光颜色随Br/I含量比的不同可由绿色调至深红色。对于CsPbBr3-xClx固溶体量子点的微晶玻璃粉,其发光颜色可由紫外调至绿色。
进一步将研磨获得的微晶玻璃粉放入电阻炉中加热至450℃后保温2小时,以使玻璃基体内部发生晶化,可显著提高CsPbBr3量子点的结晶含量(如图5所示),其发射强度随之得到提升(如图6所示)。
实施例2
将分析纯的B2O3,SiO2,ZnO,BaO,Cs2O,PbBr2,PbI2,NaI,NaBr粉体,按30B2O3:30SiO2:10ZnO:5BaO:5Cs2O:5PbBr2:5PbI2:5NaI:5NaBr(摩尔百分比)的配比精确称量后置于研钵中,混合并研磨均匀后置于氧化铝坩埚中,放入电阻炉中加热到800℃后保温0.5小时使之熔融,而后,将玻璃熔体取出并快速倒入模具中成形,得到块状前驱玻璃,最后,将获得的前驱玻璃放入电阻炉中在300℃退火以消除内应力。将得到的前驱玻璃块体置于玻璃破碎机中碾碎成小颗粒,进而将这些玻璃颗粒放入球磨机中进一步研细成微米级的微晶玻璃粉。球磨机转数为600转/分钟;球磨时间为6小时。
按照与实施例1中相同的测试方式,玻璃表面析出CsPbBr3-xIx固溶体量子点晶相,可产生发光颜色由绿色调至深红色的窄带激子发射。可将研磨获得的微晶玻璃粉放入电阻炉中加热至550℃后保温6小时,以使玻璃基体内部发生进一步晶化,可显著提高CsPbBr3-xIx量子点的结晶含量,其发射强度随之得到提升。
实施例3
将分析纯的GeO2,B2O3,ZnO,BaO,K2O,Cs2O,PbBr2,PbCl2,NaCl粉体,按50GeO2:10B2O3:5ZnO:5BaO:5K2O:5Cs2O:6PbBr2:4PbCl2:10NaCl(摩尔百分比)的配比精确称量后置于研钵中,混合并研磨均匀后置于铂金坩埚中,放入电阻炉中加热到700℃后保温1小时使之熔融,而后,将玻璃熔体取出并快速倒入模具中成形,得到块状前驱玻璃,最后,将获得的前驱玻璃放入电阻炉中在220℃退火以消除内应力。将得到的前驱玻璃块体置于玻璃破碎机中碾碎成小颗粒,进而将这些玻璃颗粒放入球磨机中进一步研细成微米级的微晶玻璃粉。球磨机转数为400转/分钟;球磨时间为8小时。
按照与实施例1中相同的测试方式,玻璃表面析出CsPbBr3-xClx固溶体量子点晶相,可产生发光颜色由紫外调至绿色的窄带激子发射。可将研磨获得的微晶玻璃粉放入电阻炉中加热至500℃后保温1小时,以使玻璃基体内部发生进一步晶化,可显著提高CsPbBr3-xClx量子点的结晶含量,其发射强度随之得到提升。
实施例4
将分析纯的TeO2,Al2O3,B2O3,ZnO,Na2O,BaO,Cs2O,PbCl2,NaCl粉体,按50TeO2:5Al2O3:10B2O3:5ZnO:5Na2O:5BaO:5Cs2O:10PbCl2:5NaCl(摩尔百分比)的配比精确称量后置于研钵中,混合并研磨均匀后置于石英坩埚中,放入电阻炉中加热到550℃后保温3小时使之熔融,而后,将玻璃熔体取出并快速倒入模具中成形,得到块状前驱玻璃,最后,将获得的前驱玻璃放入电阻炉中在250℃退火以消除内应力。将得到的前驱玻璃块体置于玻璃破碎机中碾碎成小颗粒,进而将这些玻璃颗粒放入球磨机中进一步研细成微米级的微晶玻璃粉。球磨机转数为500转/分钟;球磨时间为10小时。
按照与实施例1中相同的测试方式,玻璃表面析出CsPbCl3量子点晶相,可产生紫外窄带激子发射。可将研磨获得的微晶玻璃粉放入电阻炉中加热至400℃后保温10小时,以使玻璃基体内部发生进一步晶化,可显著提高CsPbCl3量子点的结晶含量,其发射强度随之得到提升。
实施例5
将分析纯的P2O5,SiO2,ZnO,BaO,Cs2O,PbI2,NaI粉体,按30P2O5:25SiO2:15ZnO:5BaO:5Cs2O:10PbI2:10NaI(摩尔百分比)的配比精确称量后置于研钵中,混合并研磨均匀后置于石英坩埚中,放入电阻炉中加热到650℃后保温0.5小时使之熔融,而后,将玻璃熔体取出并快速倒入模具中成形,得到块状前驱玻璃,最后,将获得的前驱玻璃放入电阻炉中在200℃退火以消除内应力。为了评估其笔迹鉴定能力,将获得的玻璃在砂纸上用不同力量进行书写。将得到的前驱玻璃块体置于玻璃破碎机中碾碎成小颗粒,进而将这些玻璃颗粒放入球磨机中进一步研细成微米级的微晶玻璃粉。球磨机转数为200转/分钟;球磨时间为6小时。
按照与实施例1中相同的测试方式,玻璃表面析出CsPbI3量子点晶相,可产生深红窄带激子发射。可将研磨获得的微晶玻璃粉放入电阻炉中加热至300℃后保温8小时,以使玻璃基体内部发生进一步晶化,可显著提高CsPbI3量子点的结晶含量,其发射强度随之得到提升。
一种无机钙钛矿量子点的固相合成方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0