

IPC分类号 : C07F7/28,C07F7/00,C08F10/00,C08F110/02,C08F210/16,C08F293/00,C08F4/645,C08F4/656,C08F4/651










专利摘要
本发明公开了一种限制构型双金属化合物及其制备方法与应用。该化合物具有双中心,不仅具有单中心催化剂的活性,而且很方便的通过调节取代基,控制两个金属中心的电子环境与空间环境,进而调控聚合物的分子量分布,生产出高分子量的、宽分布聚烯烃,并且应用于制备乙烯/1‑辛烯共聚物。本发明所述制备方法具有合成路线短,合成工艺简单、工业成本低廉的优点。
权利要求
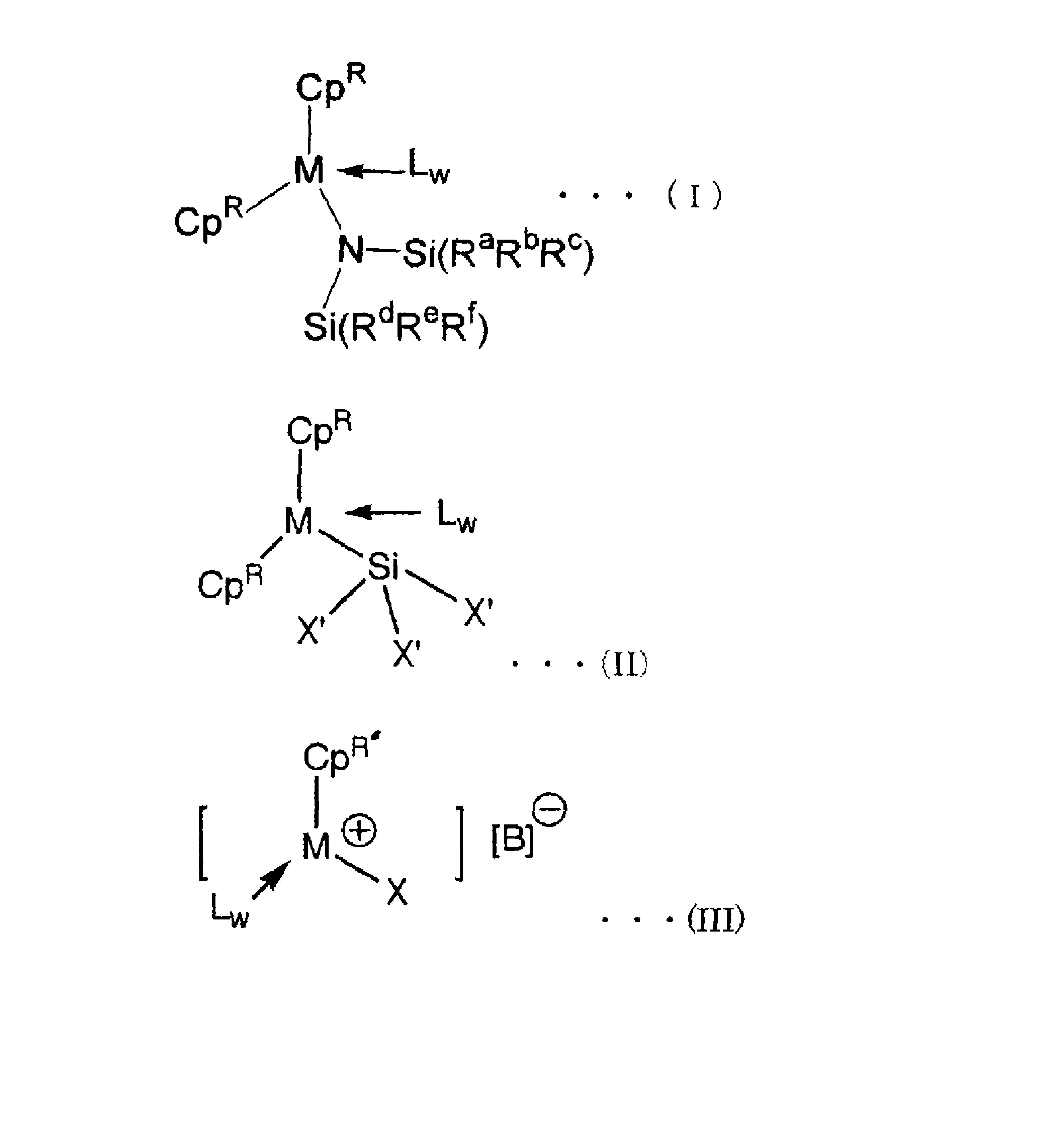
1.一种限制构型双金属化合物,其结构式如下所示:
其中:
R
R
M选自Ti或Zr。
2.一种权利要求1所述的限制构型双金属化合物,其特征在于,包含以下化合物:
[(o-O(C
[(o-O(C
[(o-O(C
[(o-(C
[(o-(C
[(o-(C
3.权利要求1或2所述的限制构型双金属化合物作为烯烃聚合反应的催化剂的应用。
4.根据权利要求3所述的应用,其特征在于,所述限制构型双金属化合物作为主催化剂,甲基铝氧烷作为助催化剂,主催化剂与助催化剂的摩尔比为1:50~2000。
5.根据权利要求4所述的应用,其特征在于,所述限制构型双金属化合物作为主催化剂,甲基铝氧烷作为助催化剂,主催化剂与助催化剂的摩尔比为1:50,聚合活性超过2.90×10
6.根据权利要求3所述的应用,其特征在于,所述限制构型双金属化合物作为催化剂,与亚胺镍、乙基锌组成催化体系,制备乙烯-1-辛烯共聚物,聚合物中1-辛烯含量1.06%~13.63%,结晶度低于50%。
7.一种权利要求1所述的限制构型双金属化合物的制备方法,其特征在于,包括以下步骤:
(1)在氩气保护下,以有机酸作为催化剂,将对苯二胺、取代水杨酸和溶剂加入反应器中,在0~90℃温度下反应1~10小时,反应结束后除去溶剂,所得固体干燥后用甲醇或乙醇重结晶,得到有机配体L;其中,对苯二胺、取代水杨醛与有机酸摩尔比为1.0:2.0~2.2:0.05~0.1;
(2)在氩气保护下,在25~80℃温度下,使配体L与Et
其中,配体L、Et
(3)将配体L的硅醚化合物溶解于二氯甲烷中,在-78~30℃温度下,滴入MCl
8.根据权利要求7所述的制备方法,其特征在于,所述取代水杨醛为水杨醛、3-甲基水杨醛、3-烯丙基水杨醛、3-氯水杨醛或3,5-二叔丁基水杨醛中的一种。
9.根据权利要求7所述的制备方法,其特征在于,所述有机酸为甲磺酸、苯磺酸、对甲基苯磺酸或三氯乙酸中的一种。
10.根据权利要求7所述的制备方法,其特征在于,步骤(1)中,所述溶剂为甲醇或乙醇;步骤(2)中,所述溶剂为正己烷或正戊烷。
说明书
技术领域
本发明涉及聚烯烃催化剂技术领域,具体涉及一种限制构型双金属化合物及其制备方法与应用。
背景技术
聚烯烃催化剂作为聚烯烃工业的核心,一直受到国内外学术界和产业界的广泛关注。Mitsui Chemicals在1998年开发的FI催化剂,作为一类新型具有高性能烯烃配位聚合的非茂金属催化剂[Weiser M S,Thomann Y,Heinz L C,et al.Polymer.2006,47(13):4505-4512],其催化剂结构中含有苯氧基亚胺结构或类似结构的配体,配体结构多,使得其空间位阻和电子效应能够在很宽的范围内进行调节,从而获得空间性能和电子性能独特的催化剂。
2002年三井化学成功开发的FI催化剂,用来工业化生产新材料单端双键低聚物。该产品分子量为2000-5000,具有好的耐热性、高结晶度、高不饱和系数、高熔点(120℃以上)等特点,也可与各种官能团和单体共聚[Kojoh S,Matsugi T,Saito J,etal.J.Chem.Lett.2001,30(8):822-823],如双键与不同单体聚合,可制备嵌段聚合物、长链分支聚合物和极性聚合物等新型聚合物。实验研究证明FI催化剂具有许多独特的化学特性,如准确控制链转移反应,催化乙烯聚合时表现出超高活性,比目前已应用于工业生产的茂金属催化剂活性高出数十倍。因此,FI催化剂的聚合性能(尤其是催化共聚)与各种配体结构之间关系的系统研究备受学者关注。
研究表明,单核催化剂得到的聚合物分子量分布狭窄,利用双金属的协同效应可以有效解决这个问题。因此,研究者进行了双金属催化剂的研制工作。Salata和Marks[Sakuma A,Weiser M S,Fujita T.Polymer Journal.2007,39(3):193-207]合成了刚性的平面结构的萘氧基胺烯烃聚合催化剂,其结构式如下:
相比于柔性桥基,萘环骨架阻止了在催化过程中金属中心的相互旋转偏离,得到高分子量的线性聚乙烯,且是单核催化剂催化活性的8倍。
美国专利US9504248报道,Sergey等人也于2012年制备了一类双水杨醛桥联亚胺的双核钛的配合物,其结构式如下:
由于亚胺中桥基和取代酚的作用使得该类催化剂比单核催化剂具有更好的热稳定性,且催化活性也很高,得到超高分子量的聚乙烯。
上述催化剂的合成路线长,原料昂贵。用对苯基桥联单核FI金属化合物得到双金属化合物是个全新的研究领域。而且通过调节取代基,可以有效控制两个金属中心的空间效应和电子效应,产生性能不同的聚烯烃。因此,使用价格低廉的原料,缩短合成路线,简化合成工艺,降低成本,提高聚合物分子量分布与制备共聚物仍然是本领域亟待解决的问题之一。
发明内容
发明目的:针对现有技术的不足,本发明的目的之一是提供一种限制构型双金属化合物。
本发明的另一目的是提供上述限制构型双金属化合物作为烯烃聚合反应的催化剂的应用。
本发明还有一个目的是提供上述限制构型双金属化合物的制备方法。
技术方案:为实现上述发明目的,本发明采用的技术方案是:
一种限制构型双金属化合物,其结构式如下所示:
其中:
R
R
M选自Ti或Zr;
用对苯基连接两个单核FI化合物,水杨醛中苯环上引入甲基、烯丙基、叔丁基以及氯等基团,得到了一种限制构型双金属化合物,该化合物的结构特征为对苯基桥连限制两个金属中心距离,通过调节取代基,可以很便利控制两个金属中心的空间和电子效应,进而更好调控聚合物分子量的分布以及共聚性能。
优选地,所述限制构型双金属化合物,包含以下化合物:
[(o-O(C6H4)CH=N)(TiCl3)]2(p-C6H4)、
[(o-O(C6H3)-3-Me)CH=N)(TiCl3)]2(p-C6H4)、
[(o-O(C6H3)-3-allyl)CH=N)(TiCl3)]2(p-C6H4)、
[(o-(C6H2)-3,5-tBu2)OCH=N)(TiCl3)]2(p-C6H4)、
[(o-(C6H2)-3,5-tBu2)OCH=N)(ZrCl3)]2(p-C6H4)、
[(o-(C6H3)-3-Cl)OCH=N)(TiCl3)]2(p-C6H4)。
所述的限制构型双金属化合物作为烯烃聚合反应以及共聚反应的催化剂的应用。
利用上述限制构型双金属化合物/甲基铝氧烷(MAO)作为催化剂的应用方式为:在乙烯压力为1.0MPa下,以甲苯或者庚烷为溶剂,加入限制构型双金属化合物的甲苯溶液,助催化剂MAO,在反应温度0~90℃下,反应0.5小时,催化乙烯聚合或者乙烯与α-烯烃共聚合反应。
优选地,所述限制构型双金属化合物作为主催化剂,甲基铝氧烷(MAO)作为助催化剂,主催化剂与助催化剂的摩尔比为1:50~2000。
进一步优选地,所述限制构型双金属化合物作为主催化剂,甲基铝氧烷(MAO)作为助催化剂,主催化剂与助催化剂的摩尔比为1:50,聚合活性超过2.90×10
所述限制构型双金属化合物作为催化剂,与亚胺镍、乙基锌组成催化体系,制备乙烯-1-辛烯共聚物,聚合物中1-辛烯含量1.06%~13.63%,结晶度低于50%。
一种所述限制构型双金属化合物的制备方法,包括以下步骤:
(1)在氩气保护下,以有机酸作为催化剂,将对苯二胺、取代水杨酸和溶剂加入反应器中,在0~90℃温度下反应1~10小时,反应结束后除去溶剂,所得固体干燥后用甲醇或乙醇重结晶,得到有机配体L;其中,对苯二胺、取代水杨醛与有机酸摩尔比为1.0:2.0~2.2:0.05~0.1;
(2)在氩气保护下,在25~80℃温度下,使配体L与Et3N和Me3SiCl在溶剂中反应3~10小时,过滤除去白色沉淀,滤液缓慢浓缩至淡黄色晶体析出,过滤得到配体L的硅醚化合物,其结构式如下所示:
其中,配体L、Et3N与Me3SiCl的摩尔比为1:2~10:2~8;
(3)将配体L的硅醚化合物溶解于二氯甲烷中,在-78~30℃温度下,滴入MCl4或CpMCl3的四氢呋喃溶液,滴加完毕后升温至25℃反应3~16小时,所得到的固体经后处理得到限制构型双金属化合物;
所述MCl4选自TiCl4和ZrCl4中的一种,所述CpMCl3选自CpTiCl3和CpZrCl3中的一种;
其中,配体L的硅醚化合物与MCl4或CpMCl3的摩尔比为1:2~2.4。
上述步骤(1)中,所述取代水杨醛为水杨醛、3-甲基水杨醛、3-烯丙基水杨醛、3-氯水杨醛或3,5-二叔丁基水杨醛中的一种。
上述步骤(1)中,所述有机酸为甲磺酸、苯磺酸、对甲基苯磺酸或三氯乙酸中的一种。
上述步骤(1)中,所述溶剂为甲醇或乙醇;上述步骤(2)中,所述溶剂为正己烷或正戊烷。
进一步优选地,上述步骤(3)中,所述固体的后处理用烷烃洗涤后,还可以用甲苯进行重结晶处理。
本发明提供的硅醚化合成方法为合成三氯化钛化合物提供新的合成路径,实践结果表明,直接使用配体是得不到目标产物的。
有益效果:(1)本发明提供了一种限制构型双金属化合物,该化合物具有双中心,空间构型稳定,使用苯环作为连接官能团限制了金属中心距离;另一方面,在空间上金属中心在苯环两侧,完全不影响烯烃插入,因此具有良好的催化活性和共聚能力。
(2)本发明提供的限制构型双金属化合物的制备方法,采用对苯二胺与取代水杨醛制备双官能团配体,硅醚化后进一步与金属络合,得到双中心化合物,具有合成路线短,合成工艺简单,易于分离纯化,产品收率高,工业成本低廉的优点。
(3)本发明制得的限制构型双金属化合物,其不仅保留了单中心催化剂的活性,而且很方便的通过调节取代基,控制两个金属中心的空间效应和电子效应,进而调控聚合物的分子量及其分布,可生产出宽分布的聚烯烃以及共聚物。
(4)限制构型双金属化合物/MAO的催化剂体系,在较低的助催化剂比例条件下,依然对乙烯聚合以及共聚具有较高活性,聚合物的中1-辛烯插入率可达13.63mol%,聚合物呈现嵌段共聚物结构。
附图说明
图1为实施例21聚合物
图2为实施例22聚合物
图3为实施例21、22制备的乙烯/1-辛烯共聚物的DSC曲线;
图4为乙烯/1-辛烯共聚物中“软段”结构图;
图5为实施例21制得的乙烯/1-辛烯嵌段共聚物结构。
具体实施方式
下面通过附图对本发明技术方案进行详细说明,但是本发明的保护范围不局限于所述实施例。
实施例1
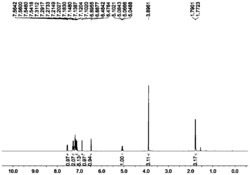
本实施例提供了一种限制构型双金属化合物,该络合物C1的分子式为:[(o-O(C6H4)CH=N)(TiCl3)]2(p-C6H4)。
络合物C1的合成路线如下:
络合物C1的具体制备过程为:
(1)氩气保护下,在装有磁力搅拌器的250mL三颈烧瓶上接一个回流冷凝管,室温下,向瓶中加入水杨醛(4.88g,0.04mol)、对苯二胺(2.16g,0.02mol)和120mL乙醇,甲基磺酸1mL,加热回流6-8h,有橙黄色固体产生。停止搅拌,恢复室温,抽滤,固体用乙醇洗涤,干燥,得到橙黄色固体L1,5.82g,产率92.0%。
(2)无水无氧条件下,在装有磁力搅拌器的250mL三颈烧瓶上接一个回流冷凝管,室温下,向瓶中加入L1(0.98g,3.0mmol),用180mL正己烷溶解,再加入三乙胺(2.43g,24.0mmol),三甲基氯硅烷(1.96g,18.0mmol),加热回流过夜,有白色絮状固体产生。停止反应,逐渐恢复至室温,白色固体沉淀,溶液为淡黄色,压滤,滤液用真空缓慢浓缩,期间有淡黄色晶体析出,保留少量滤液,压滤,固体抽干,直接用于下一步反应。
(3)同样在无水无氧条件下,在装有磁力搅拌器的100mL Schlenk瓶上配一个恒压滴液漏斗,上述固体(1.11g,2.4mmol)用20mL二氯甲烷溶解后,在液氮乙醇浴条件下,滴加到40mL溶解的TiCl4(4.8mmol)溶液中。滴加结束后,逐渐恢复至室温,持续搅拌反应一天一夜,有暗红色固体产生。反应结束后,静置沉淀,压滤,固体各用二氯甲烷和乙醚洗涤2~3次,抽干,得到暗红色产品C1,产率82.5%。
实施例2
本实施例提供了一种限制构型双金属化合物,该络合物C2的分子式为:[(o-O(C6H3)-3-Me)CH=N)(TiCl3)]2(p-C6H4)。
络合物C2的制备过程与实施例1中络合物C1的制备过程相同,区别仅在于用3-烯丙基水杨醛代替水杨醛,苯磺酸代替甲磺酸。产率76.0%。
实施例3
本实施例提供了一种限制构型双金属化合物,该络合物C3的分子式为:[(o-O(C6H3)-3-allyl)CH=N)(TiCl3)]2(p-C6H4)。
络合物C3的制备过程与实施例1中络合物C1的制备过程相同,区别仅在于用3-烯丙基水杨醛代替水杨醛,对甲基苯磺酸代替甲磺酸。产率59.6%。
实施例4
本实施例提供了一种限制构型双金属化合物,该络合物C4的分子式为:[(o-(C6H2)-3,5-tBu2)OCH=N)(TiCl3)]2(p-C6H4)。
络合物C4的制备过程与实施例1中络合物C1的制备过程相同,区别仅在于用3,5-二叔丁基水杨醛代替水杨醛,三氯乙酸代替甲磺酸。得到棕色固体,产率70.1%。
实施例5
本实施例提供了一种限制构型双金属化合物,该化合物(络合物C5)的分子式为:[(o-(C6H2)-3,5-tBu2)OCH=N)(ZrCl3)]2(p-C6H4)。
络合物C5的制备过程与实施例4中络合物C4的制备步骤(1)相同,制得化合物L5。
无水无氧条件下,在装有磁力搅拌器的50mL Schlenk瓶中加入L5(0.54g,1.0mmol),用30mL THF溶解,冰水浴条件下,加入NaH(0.08g,2.0mmol),有气泡产生,恢复室温反应5h。静置沉降,压滤,得到橙色透明液。另取CpZrCl3(0.60g,2mmol),用50mL THF溶解于100mL Schlenk瓶中,冰水浴条件下,向其滴加上述的配体钠盐。滴加结束后,逐渐恢复至室温,持续搅拌反应一天一夜,静置沉淀,压滤,滤液抽干,重结晶,得到黄色固体,产率51.6%。
实施例6
本实施例提供了一种限制构型双金属化合物,该化合物(络合物C6)的分子式为:[(o-(C6H3)-3-Cl)OCH=N)(TiCl3)]2(p-C6H4)
络合物C6的制备过程与实施例1中络合物C1的制备过程相同,区别仅在于用3-氯水杨醛代替水杨醛。得到棕黄色固体,产率81.7%。
实施例7
本实施例提供了将实施例1制得的络合物C1作为催化剂催化乙烯聚合的应用。
均相催化乙烯聚合包括以下步骤:
将配有磁力搅拌子和导气管的250mL高压釜用乙烯气体置换3次,在氮气保护下,依次加入甲苯、助催化剂MAO 6.7mL(1.50M)[Al/M=1000],5.0μmol络合物C1,控制总体积为100mL,通入乙烯气体,在60℃下开始聚合反应,维持乙烯压力1.0MPa,搅拌反应30min,关闭气瓶,泄压,然后用10%的盐酸乙醇终止反应。将聚合物转移到烧杯中,静置过夜,过滤并用乙醇充分洗涤聚合物,在60℃下真空干燥至恒重,称量聚合物质量,计算后得出催化剂聚合活性为7.62×10
实施例8
本实施例提供了实施例2制得的络合物C2作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C2代替C1。计算后得出催化剂聚合活性8.72×10
实施例9
本实施例提供了实施例3制得的络合物C3作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C3代替C1。计算后得出催化剂聚合活性1.17×10
实施例10
本实施例提供了实施例4制得的络合物C4作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C4代替C1。计算后得出催化剂聚合活性5.61×10
实施例11
本实施例提供了实施例5制得的络合物C5作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C5代替C1。计算后得出催化剂聚合活性1.23×10
实施例12
本实施例提供了实施例6制得的络合物C6作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C6代替C1。计算后得出催化剂聚合活性1.56×10
实施例13
本实施例提供了实施例3制得的络合物C3作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C3代替C1。聚合条件:反应温度30℃,乙烯压力1.0MPa,其他条件与实施例7相同。计算后得出催化剂聚合活性1.47×10
实施例14
本实施例提供了实施例3制得的络合物C3作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C3代替C1。聚合条件:反应温度90℃,其他条件与实施例7相同。计算后得出催化剂聚合活性1.15×10
实施例15
本实施例提供了实施例3制得的络合物C3作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C3代替C1。聚合条件:反应温度90℃,Al/Ti=500,其他条件与实施例7相同。计算后得出催化剂聚合活性5.16×10
实施例16
本实施例提供了实施例4制得的络合物C4作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C4代替C1。聚合条件:反应温度30℃,乙烯压力1.0MPa,其他条件与实施例7相同。计算后得出催化剂聚合活性1.18×10
实施例17
本实施例提供了实施例4制得的络合物C4作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C4代替C1。聚合条件:反应温度30℃,Al/M=300,其他条件与实施例7相同。计算后得出催化剂聚合活性7.83×10
实施例18
本实施例提供了实施例4制得的络合物C4作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C4代替C1。聚合条件:反应温度30℃,Al/Ti=50,乙烯压力1.5MPa,其他条件与实施例7相同。计算后得出催化剂聚合活性5.89×10
实施例19
本实施例提供了实施例5制得的络合物C5作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C5代替C1。聚合条件:反应温度90℃,其他条件与实施例7相同。计算后得出催化剂聚合活性2.10×10
实施例20
本实施例提供了实施例5制得的络合物C5作为催化剂催化乙烯聚合的应用。
聚合操作与实施例7相同,以C5代替C1。聚合条件:反应温度90℃,Al/Zr=50,其他条件与实施例7相同。计算后得出催化剂聚合活性2.43×10
实施例21
本实施例提供了实施例5制得的络合物C5作为催化剂制备乙烯/1-辛烯共聚物的应用。使用已知化合物N1,结构式如下:
均相催化乙烯聚合包括以下步骤:
将配有磁力搅拌子和导气管的250mL高压釜用乙烯气体置换3次,在氮气保护下,依次加入甲苯、助催化剂MAO 4.0mL(1.50M)[Al/M=300],5.0μmol络合物C5,10μmol络合物N1,0.5mL的ZnEt2。控制总体积为100mL,通入乙烯气体,在50℃下开始聚合反应,维持乙烯压力1.0MPa,搅拌反应30min,关闭气瓶,泄压,然后用10%的盐酸乙醇终止反应。将聚合物转移到烧杯中,静置过夜,过滤并用乙醇充分洗涤聚合物,在60℃下真空干燥至恒重,称量聚合物质量,计算后得出催化剂聚合活性为8.14×10
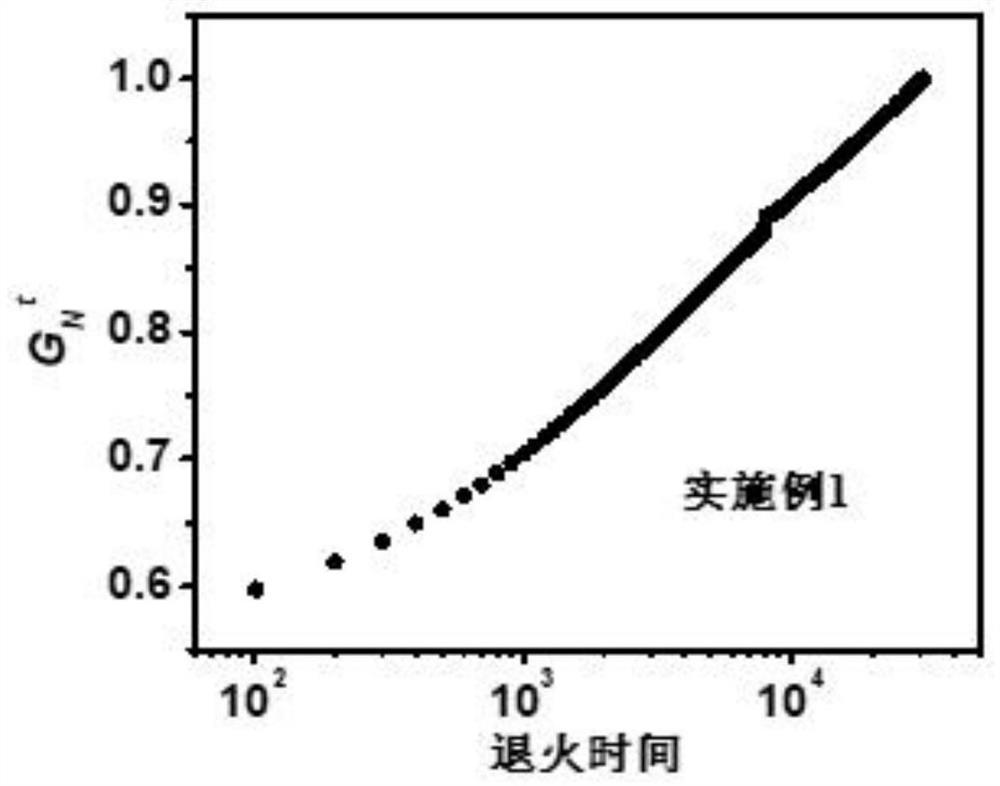
从图1可以看出,共聚物中没有OOO片段,也没有EOO和OOE片段以及EOEOE片段,只有经典的EOE结构单元,这表明该聚合物中,1-辛烯是随机分布的。
实施例22
本实施例提供了实施例5制得的络合物C5作为催化剂制备乙烯/1-辛烯共聚物的应用。
除了不加入ZnEt2,其他条件同实施例21。聚合结果:聚合活性为1.89×10
从图2可以看出,聚合物中没有OOO片段,表明聚合物中没有连续的聚1-辛烯结构单元,最主要的结构单元是EOE,但是含有EOO和OOE结构片段以及EOEOE片段,这应该是由于Et2Zn引起的链穿梭的结果。
从图3可以看出,与实施例22相比,加入Et2Zn以后得到的聚合物(实施例21)其结构明显有变化,呈现软硬段交替结构,因此DSC出现双峰分布。
表1络合物C5-N1催化乙烯-1-辛烯聚合物热性能参数
从实施例结果看,该类催化剂能够催化乙烯聚合得到高分子量的、宽分布的聚乙烯。此外,从表1中聚合物表征参数可以看出,本发明合成的催化剂与N1-Et2Zn组合,可以有效催化乙烯与1-辛烯共聚,其中,乙烯与1-辛烯共聚物片段结构见附图4,图中左边是1-辛烯随机分布,右边是链穿梭引起的EOO、OOE、OEO结构单元。
这种催化体系制备得到的乙烯/1-辛烯共聚物的结构为聚乙烯与乙烯/1-辛烯共聚物的嵌段聚合物(见附图5),其中“硬段”部分是由FI催化剂为主所形成的线性聚乙烯构成,而“软段”部分则是由附图4为主要结构的乙烯/1-辛烯共聚物构成。
如上所述,尽管参照特定的优选实施例已经表示和表述了本发明,但其不得解释为对本发明自身的限制。在不脱离所附权利要求定义的本发明的精神和范围前提下,可对其在形式上和细节上作出各种变化。
一种限制构型双金属化合物及其制备方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0