

IPC分类号 : C07J1/00,C07J31/00,C07J43/00,A61K31/569,A61K31/58,A61P25/24







专利摘要
本发明公开了一种抗抑郁化合物,所述的抗抑郁化合物,为式I的结构。所述的抗抑郁化合物作为抗抑郁保健食品的唯一活性成分或者活性成分之一。所述的抗抑郁药物为液体制剂、固体制剂、喷剂或者气雾剂等。本发明抗抑郁化合物在制备抗抑郁药物和抗抑郁保健食品中的应用,该抗抑郁化合物与已知抗抑郁药的化学结构均不相同。该抗抑郁化合物具有显著的抗抑郁活性,并且,未见有明显毒副反应,有利于市场化大规模推广利用,具备广阔的应用前景。
权利要求
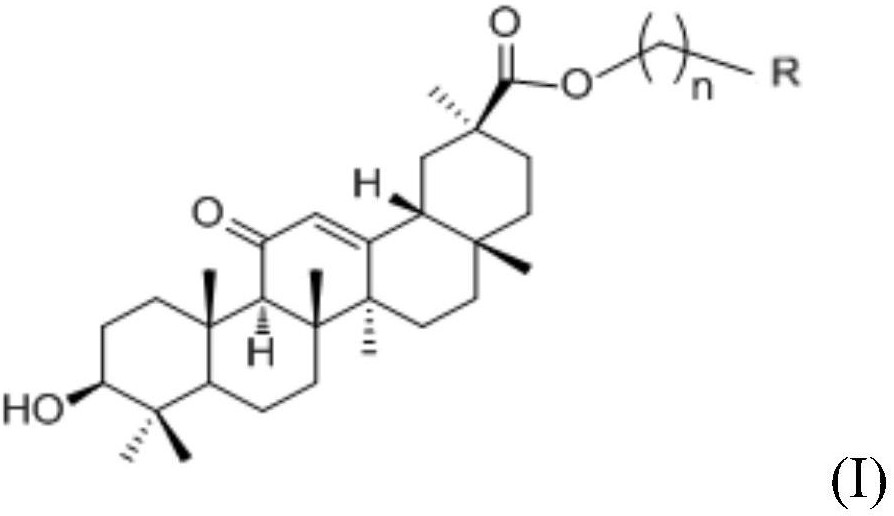
1.一种抗抑郁化合物,其特征在于,所述的抗抑郁化合物,为式I的结构:
取代基R1为羟基、硫酸酯或醋酸酯;
取代基R2为羟基或酯基;
取代基R为乙酰基或CH3CHR3,其中,CH3CHR3中的取代基R3为羟基或酯基。
2.根据权利要求1所述的一种抗抑郁化合物,其特征在于,所述的取代基R2中的酯基为醋酸酯、(Z)2-甲基-2-丁烯酸酯、烟酸酯、肉桂酸酯、(N-甲基)邻氨基苯甲酸酯、苯甲酸酯、对羟基苯甲酸酯或者(2E)-3,4-二甲基-2-烯-戊酸酯。
3.根据权利要求1所述的一种抗抑郁化合物,其特征在于,所述的取代基R3中的酯基为醋酸酯、(Z)2-甲基-2-丁烯酸酯、烟酸酯、肉桂酸酯、(N-甲基)邻氨基苯甲酸酯或者苯甲酸酯。
4.根据权利要求1所述的一种抗抑郁化合物,其特征在于,所述的抗抑郁化合物中,取代基R1为羟基、硫酸酯或醋酸酯;取代基R2为(Z)2-甲基-2-丁烯酸酯,取代基R为CH3CHR3,且取代基R3为(Z)2-甲基-2-丁烯酸酯,或者,取代基R2为(N-甲基)邻氨基苯甲酸酯且取代基R为乙酰基。
说明书
技术领域
本发明涉及医药技术领域,具体涉及一种抗抑郁化合物在制备抗抑郁药物和抗抑郁保健食品中的应用。
背景技术
抑郁症是一种高发的精神障碍性疾病,当前临床传统的抗抑郁药物主要是针对抑郁症患者脑内单胺类神经递质,虽然是有效安全的,但其主要缺陷是:起效慢(需要数周甚至数个月才能显效减缓症状)、治疗产生抵抗(难治性抑郁症)、会复发,因此,临床迫切需要开发速效及针对难治性抑郁症的抗抑郁药物。
根据抑郁症的发病机制围绕神经可塑性、神经发生、下丘脑-垂体-肾上腺(HPA)轴等后续神经系统适应性改变,目前抗抑郁药物的靶标主要有[Newman DJ,Cragg GM.Natural products as sources of new drugs over the 30years from 1981to 2010.J Nat Prod,2012,75(3):311-35.]:①作用于单胺能系统(提高抑郁症患者脑内神经递质中的生物胺:5羟色胺,5-HT;去甲肾上腺素能,NE等)如有5-HT重摄取抑制剂(SSRIs)、NE再摄取抑制剂(SNRIs)及多巴胺调节剂;②作用于谷氨酸受体,如NMDA受体拮抗剂、AMPA受体调节剂;③作用于神经肽类受体如神经激肽(NK)受体拮抗剂、促肾上腺皮质激素(CRH)受体拮抗剂;④作用于糖皮质激素受体(GR)如糖皮质激素受体拮抗剂。但是当前临床传统的抗抑郁药物主要是针对抑郁症患者脑内单胺类神经递质,虽然是有效安全的,但其主要缺陷是起效慢(需要数周甚至数个月才能显效减缓症状)、治疗产生抵抗(难治性抑郁症,treatment-resistant depression)、会复发,因此临床迫切需要开发速效及针对难治性抑郁症的抗抑郁药物[Mathew SJ,Manji HK,Charney DS.Novel drugs and therapeutic targets for severe mood.Neuropsychopharmacology 2008:1-13.2.Cryan JF,OLeary OF.A glutamate pathway to faster-acting antidepressants?Science 2010;329:913.]。
近年来基于非单胺能生物系统(单胺能系统外)的新机制抗抑郁药物研究引起广泛了的重视[Berton O,Nestler EJ.New approaches to antidepressant drug discovery:beyond monoamines.Neurosience 2006;7:137-151.],世界各大制药企业在新型抗抑郁药物的开发掀起浪潮,发现大量具有抗抑郁作用的新化合物,褪黑激素受体激动剂阿戈美拉丁2009年在欧洲上市、美国FDA 2009年批准促肾上腺皮质激素释放激素受体拮抗剂喹硫平作为抗抑郁药。还有许多治疗重症抑郁症(Major Depression)的药物,有氨基酸神经递质受体拮抗剂(如NMDA拮抗剂),神经多肽类拮抗剂(CRF-1、NK-1拮抗剂),糖皮质激素受体拮抗剂(GR拮抗剂)进入美国FDA II和III期临床试验[孟秀君,曲蕾,马燕等,新型抗抑郁药物的研究进展中国新药杂志2011;20:1766-1774.]。
C21甾体化合物在植物界广泛分布,尤以萝藦科植物中分布最为普遍,从植物中分离出的C21甾体化合物都是以孕甾烷或其异构体为基本骨架,在植物中主要以苷元与糖结合成苷的形式存在,糖苷在酸性条件下可以水解成次级苷或苷元。有关植物来源C21甾体苷的抗抑郁作用近年来已有文献报道:中国专利有关于几种植物C21甾体配糖体(混合物)治疗和预防抑郁药物中的应用[一种C21甾体配糖体在制药中的应用.CN 1634097A.];文献报道了从萝藦科植物耳叶牛皮消中分离到三个具有抗抑郁作用的C21甾体苷[Yang QX,Ge YC,Huang XY,et.al.,Cynanauriculoside C-E,three new antidepressant pregnane glycosides from Cynanchum auriculatum.Phytochemistry Letters 2011;4:170-175.]。
发明内容
本发明提供了一种抗抑郁化合物,它可在制备抗抑郁药物和抗抑郁保健食品中的应用,该抗抑郁化合物与已知抗抑郁药的化学结构均不相同。
一种抗抑郁化合物,所述的抗抑郁化合物,为式I的结构:
取代基R1为羟基、硫酸酯或醋酸酯;
取代基R2为羟基或酯基;
取代基R为乙酰基或CH3CHR3,其中,CH3CHR3中的取代基R3为羟基或酯基。
取代基R2为酯基时,其中的酯基又可为如下基团之一:即所述的取代基R2中的酯基为醋酸酯、(Z)2-甲基-2-丁烯酸酯、烟酸酯、肉桂酸酯、(N-甲基)邻氨基苯甲酸酯、苯甲酸酯、对羟基苯甲酸酯或者(2E)-3,4-二甲基-2-烯-戊酸酯。
取代基R3为酯基时,其中的酯基又可为如下基团之一:即所述的取代基R3中的酯基为醋酸酯、(Z)2-甲基-2-丁烯酸酯、烟酸酯、肉桂酸酯、(N-甲基)邻氨基苯甲酸酯或者苯甲酸酯。
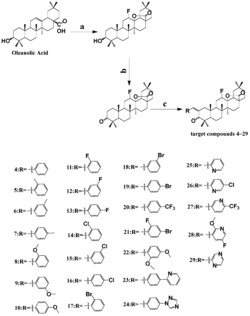
上述这些化合物的取代基团和名称见表1。
表1肉珊瑚苷元及其衍生物、去乙酰萝藦苷元及其衍生物
例如,化合物1为肉珊瑚苷元:取代基R1=OH,取代基R2=OH,取代基R为CH3CHR3,其中取代基R3=OH;
再例如化合物18为去乙酰萝藦苷元:取代基R1=OH,取代基R2=OH,取代基R为乙酰基(CH3CO);
再例如,化合物19为萝藦苷元:取代基R1=OH,取代基R2=醋酸酯,取代基R为乙酰基(CH3CO);
再例如,化合物20为告达庭:取代基R1=OH,取代基R2=(2E)-3,4-二甲基-2-烯-戊酸酯,取代基R为乙酰基(CH3CO)。
作为优选,所述的抗抑郁化合物为表1中的化合物3、化合物23、化合物25或者化合物26。所述的抗抑郁化合物中,取代基R1为羟基、硫酸酯或醋酸酯;取代基R2为(Z)2-甲基-2-丁烯酸酯,取代基R为CH3CHR3,且取代基R3为(Z)2-甲基-2-丁烯酸酯,或者,取代基R2为(N-甲基)邻氨基苯甲酸酯且取代基R为乙酰基(CH3CO)。化合物3、化合物23为新的化合物,化合物3、化合物23对应的硫酸酯或醋酸酯衍生物也为新的化合物。
本发明具有式I结构通式所示的天然化合物及其修饰得到的衍生物,以及二者的组合即可用作药物制剂或保健食品活性成分,或活性成分之一,采用制药和食品领域公认的方法制备成各种剂型、如液体制剂(注射剂、混悬剂、乳剂、溶液剂、糖浆剂等)、固体制剂(片剂、胶囊剂、颗粒剂、冲剂等)、喷剂、气雾剂等。本发明的药物可经注射(静脉注射、静脉滴注、肌肉注射、腹腔注射、皮下注射)和口服、舌下给药、粘膜透析、经皮给药等给药途径进行抑郁症的治疗。即所述的抗抑郁化合物作为抗抑郁药物的唯一活性成分或者活性成分之一,所述的抗抑郁化合物作为抗抑郁保健食品的唯一活性成分或者活性成分之一。所述的抗抑郁药物为液体制剂、固体制剂、喷剂或者气雾剂等。所述的液体制剂为注射剂、混悬剂、乳剂、溶液剂或糖浆剂等。所述的固体制剂为片剂、胶囊剂、颗粒剂或冲剂等。
本发明所说的天然化合物(即式I的结构的抗抑郁化合物)是指从植物中提取的,特别是指从萝藦科植物黑鳗藤(Stephanotis mucronata(Blanco)Merr.)、泰山白首乌(Cynanchum bungei Decne.)以及青阳参(Cynanchum Otophyllum Schneid.)中提取的,符合上述化学结构通式的化合物。特别是从萝藦科植物黑鳗藤的茎或根部、萝藦科植物泰山白首乌的根部以及萝藦科植物青阳参的根部中提取。
本发明所说的从植物中提取,是本领域工作人员都能够掌握和采用的方法:用含水1%-90%(体积百分数,下同)的低级醇(甲醇、乙醇),或含水1%-90%的丙酮,或水饱和的丁酮、水饱和的乙酸乙酯、水饱和的氯仿、水饱和的二氯甲烷或者水饱和的正丁醇,在室温条件下(如0℃到30℃之间)或者在加热条件下(30℃以上,最高到溶剂沸点温度)下,从植物,特别是从黑鳗藤的茎或根部,泰山白首乌的根部以及青阳参的根部中溶出并制成含有上述化学结构通式的化合物的提取物。
本发明所说的符合上述化学结构通式的化合物(即式I的结构的抗抑郁化合物),是指从上述提取物中,用本领域工作人员都能够掌握和采用的柱层析方法(柱子的填料为硅胶,或辛烷基烷化硅胶,或十八烷基烷化硅胶,或葡聚糖凝胶)分离纯化,并经过波谱解析识别和确定了结构的天然来源的孕甾烷类化合物。
本发明所说的对天然来源的孕甾烷类化合物开展的化学反应和结构修饰,是指本领域工作人员都能够掌握和采用的常规化学方法,包括对天然来源的孕甾烷类化合物纯品或其混合物实施弱酸水解,断开糖链,得到3位或其它连糖位置为游离羟基的多氧孕甾酯化合物的方法;也包括用碱水解,除去原有酯基,得到含有多个羟基的衍生物的方法;也包括对天然来源的孕甾烷类化合物3位羟基进行硫酸酯化或醋酸酯化。
本发明式I结构的抗抑郁化合物用作药物时,可以直接使用,或者以药物组合物的形式使用。即所述的抗抑郁药物为药物组合物,该药物组合物含有0.1-99%(wt),优选为0.5-90%(wt)的抗抑郁化合物,其余为药物学上可接受的,对人和动物无毒和惰性的可药用载体和/或赋形剂。
所述的药用载体或赋形剂是一种或多种固体、半固体和液体稀释剂、填料以及药物制品辅剂。将本发明的药物组合物以单位体重服用量的形式使用。肉珊瑚苷元及其衍生物的组合物采用制药和食品领域公认的方法制备成各种剂型、如液体制剂(注射剂、混悬剂、乳剂、溶液剂、糖浆剂等)、固体制剂(片剂、胶囊剂、颗粒剂、冲剂等)、喷剂、气雾剂等。本发明的药物可经注射(静脉注射、静脉滴注、肌肉注射、腹腔注射、皮下注射)和口服、舌下给药、粘膜透析、经皮给药等给药途径进行抑郁症的治疗。
与现有技术相比,本发明具有如下优点:
本发明抗抑郁化合物在制备抗抑郁药物和抗抑郁保健食品中的应用,该抗抑郁化合物与已知抗抑郁药的化学结构均不相同。该抗抑郁化合物具有显著的抗抑郁活性,并且,未见有明显毒副反应,有利于市场化大规模推广利用,具备广阔的应用前景。
具体实施方式
实施例1:肉珊瑚苷元的制备和结构鉴定
泰山白首乌5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,得到乙醇提取物,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇溶液(即硫酸溶于甲醇形成硫酸甲醇溶液,其中硫酸甲醇溶液中硫酸的浓度为0.2mol/L)70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元。总苷元180g上硅胶柱层析,以二氯甲烷-甲醇系统(二氯甲烷与甲醇体积比100:0→40:60)梯度洗脱,得到Fr1-Fr8。Fr8 15g经Rp-18(反向十八烷基烷化硅胶)反复柱层析,甲醇-水系统洗脱(甲醇-水系统中甲醇的体积百分比53%→60%),TLC(薄层色谱法)检测,合并相同部分,甲醇重结晶得到肉珊瑚苷元2.3g。
肉珊瑚苷元,C21H34O6,无色针状结晶(甲醇),熔点151-153℃,254-257℃(双熔点)。ESI-MS(positive)m/z:405.1[M+Na]+.13C NMR(C5D5N,125MHz):38.9(C-1),31.7(C-2),70.39(C-3),43.02(C-4),139.69(C-5),118.55(C-6),33.74(C-7),73.73(C-8),44.13(C-9),36.88(C-10),28.72(C-11),71.18(C-12),58.2(C-13),88.43(C-14),34.09(C-15),34.88(C-16),88.51(C-17),10.84(C-18),18.06(C-19),72.64(C-20),17.32(C-21).1H NMR(C5D5N,500MHz):δ3.95(1H,m,H-3),5.45(1H,br s,H-6),3.97(1H,m,H-12),1.99(3H,s,H-18),1.49(3H,s,H-19),4.48(1H,m,H-20),1.54(3H,d,J=6.0Hz,H-21)。经光谱数据分析和理化性状测定与文献报道[Warashina T,Noro T.Steroidal glycosides from Cynanchum caudatum.Phytochemistry 1995;39(1):199-204.]的肉珊瑚苷元完全一致。
实施例2:去乙酰萝藦苷元的制备和结构鉴定
泰山白首乌5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元。总苷元180g上硅胶柱层析,以二氯甲烷-甲醇系统(100:0→40:60)梯度洗脱,得到Fr1-Fr8。Fr8 15g经Rp-18反复柱层析,甲醇-水系统洗脱(53%→60%),TLC检测,合并相同部分,甲醇重结晶,得到去乙酰萝藦苷元675mg。
去乙酰萝藦苷元,C21H32O6,无色针晶(甲醇)。IR(KBr):3510,1690cm-1。ESI-MS(positive)m/z:403.1[M+Na]+。13C NMR(C5D5N,125MHz):39.0(C-1),31.9(C-2),71.4(C-3),43.2(C-4),140.1(C-5),118.6(C-6),34.0(C-7),74.2(C-8),44.8(C-9),37.2(C-10),29.3(C-11),68.8(C-12),60.2(C-13),89.2(C-14),34.9(C-15),32.6(C-16),92.4(C-17),9.2(C-18),18.3(C-19),209.4(C-20),27.7(C-21)。1H NMR(C5D5N,500MHz):δ3.93(1H,m,H-3),5.42(1H,br s,H-6),1.96(3H,s,H-18),1.49(3H,s,H-19),3.98(1H,dd,J=11.5,4.0Hz,H-12),2.68(3H,s,H-21)。经光谱数据分析和理化性状测定与文献报道[Ye YP,Li XY,Sun HX,Chen FY,Pan YJ.Immunomodulating Steroidal Glycosides from the Roots of Stephanotis mucronata.Helvetica Chimica Acta2004;87:2378-2384]的去乙酰萝藦苷元完全一致。
实施例3:萝藦苷元的制备和结构鉴定
泰山白首乌5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元。总苷元180g上硅胶柱层析,以二氯甲烷-甲醇系统(100:0→40:60)梯度洗脱,得到Fr1-Fr8。Fr6 10g上Rp-18反复柱层析,甲醇-水系统洗脱(53%→60%),TLC检测,合并相同部分,甲醇重结晶得到萝藦苷元1.6g。
萝藦苷元,C23H34O7,无色针晶(甲醇)。IR(KBr):3510,1690cm-1。ESI-MS(positive)m/z:445.1[M+Na]+。13C NMR(C5D5N,125MHz):39.0(C-1),31.8(C-2),71.3(C-3),43.1(C-4),140.1(C-5),118.3(C-6),33.6(C-7),74.2(C-8),44.3(C-9),37.2(C-10),24.7(C-11),73.4(C-12),57.7(C-13),89.3(C-14),34.5(C-15),32.6(C-16),92.2(C-17),10.2(C-18),18.1(C-19),210.0(C-20),27.4(C-21),169.7(C-1′),20.6(C-2′)。1H NMR(C5D5N,500MHz):δ1.43(3H,s,H-19),2.51(3H,s,H-21),1.97(3H,s,H-18),5.00(1H,dd,J=11.5,4.0Hz,H-12),3.91(1H,m,H-3),5.35(1H,br s,H-6),2.10(1H,s,H-2′)。经光谱数据分析和理化性状测定与文献报道[Ye YP,Li XY,Sun HX,Chen FY,Pan YJ.Immunomodulating Steroidal Glycosides from the Roots of Stephanotis mucronata.Helvetica Chimica Acta 2004;87:2378-2384]的萝藦苷元完全一致。
实施例4:告达庭的制备和结构鉴定
泰山白首乌5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元。总苷元180g上硅胶柱层析,以二氯甲烷-甲醇系统(100:0→40:60)梯度洗脱,得到Fr1-Fr8。Fr3 23g经Rp-18反复柱层析,甲醇-水系统洗脱(55%→60%),TLC检测,合并相同部分,甲醇水重结晶得到告达庭5.2g。
告达庭,C28H42O7,无色杆状晶(甲醇-水)。ESI-MS(positive)m/z:513.1[M+Na]+。13C NMR(DMSOd6,125MHz):38.54(C-1),32.19(C-2),71.97(C-3),39.68(C-4),138.92(C-5),119.03(C-6),34.25(C-7),73.58(C-8),43.57(C-9),36.73(C-10),24.31(C-11),75.84(C-12),57.23(C-13),88.87(C-14),33.48(C-15),28.70(C-16),91.53(C-17),10.51(C-18),18.04(C-19),209.18(C-20),27.38(C-21),165.14(C-1'),113.59(C-2'),165.07(C-3'),37.61(C-4'),21.27(C-5'),21.08(C-6',),16.44(C-7')。1H NMR(DMSO d6,500MHz):δ3.88(1H,m,H-3),5.23(1H,br s,H-6),4.34(1H,dd,J=11.5,4.0Hz,H-12),2.04(3H,s,H-18),1.31(3H,s,H-19),2.50(3H,s,H-21),5.48(1H,s,H-2′),1.02(3H,d,J=7.0Hz,H-5′),1.00(3H,d,J=6.5Hz,H-6′),2.09(3H,s,H-7′)。经硅胶薄层色谱与高效液相色谱分析与告达庭对照品完全一致,并经光谱数据分析与文献报道[Zhang RS,Ye YP,Shen YM,Liang HL.Two new cytotoxic C-21steroidal glycosides from the root of Cynanchum auriculatum Tetrahedron 2000,56(24):3875-3879]的告达庭完全一致。
实施例5:青阳参苷元的制备和结构鉴定
青阳参5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇溶液70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元。总苷元165g上硅胶柱层析,以二氯甲烷-甲醇系统(100:0→45:55)梯度洗脱,得到流份Fr1-Fr3。Fr2 52g再经Rp-18反复柱层析,甲醇-水系统洗脱,得到白色固体粉末青阳参苷元5.1g。
青阳参苷元,C28H36O8,白色无定形粉末。ESI-MS(positive)m/z:523.1[M+Na]+。13C NMR(C5D5N,125MHz):39.51(C-1),32.31(C-2),71.92(C-3),43.56(C-4),140.65(C-5),118.82(C-6),35.16(C-7),74.76(C-8),44.84(C-9),37.72(C-10),25.53(C-11),73.74(C-12),58.75(C-13),89.91(C-14),33.51(C-15),34.24(C-16),92.83(C-17),11.18(C-18),18.67(C-19),210.17(C-20),28.13(C-21),15.73(C-1′),122.34(C-2′),132.75(C-3′,7′),116.52(C-4′,6′),163.91(C-5′)。1H NMR(C5D5N,500MHz):δ1.29(3H,s,H-19),2.01(3H,s,H-18),2.33(3H,s,H-21),3.74(1H,m,H-3),4.92(1H,dd,J=11.5,4.0Hz,H-12),5.26(1H,br s,H-6),7.14(2H,d,H-4′,6′),8.20(2H,d,H-3′,7′)。经硅胶薄层色谱与高效液相色谱分析与青阳参苷元对照品完全一致,并经光谱数据分析与文献报道[Ma XX,Jiang FT,Yang QX,Liu XH,Zhang YJ,Yang CR.New pregnane glycosides from the roots of Cynanchum otophyllum.Steroids2007,72:778-786]的青阳参苷元完全一致。
实施例6:12-O-乙酰基-20-O-(N-甲基)邻氨基苯甲酰基肉珊瑚苷元的制备与结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇溶液(即硫酸溶于甲醇形成硫酸甲醇溶液,其中硫酸甲醇溶液中硫酸的浓度为0.2mol/L)70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr1 16.7g,Fr2 13.0g,Fr3 14.5g。Fr1 16.7g经Rp-18(反向十八烷基烷化硅胶)柱层析,甲醇-水系统洗脱(甲醇-水系统中甲醇的体积百分比50%→60%),TLC(薄层色谱法)检测,合并相同部分,甲醇重结晶得到12-O-乙酰基-20-O-(N-甲基)邻氨基苯甲酰基肉珊瑚苷元7.8g。
12-O-乙酰基-20-O-(N-甲基)邻氨基苯甲酰基肉珊瑚苷元,C30H42NO9,无色针晶(甲醇)。HR-ESI-MS:580.2871([C31H43NO8+Na]+;计算值:580.2886)。13C NMR(C5D5N,125MHz):38.6(C-1),30.8(C-2),71.8(C-3),41.9(C-4),139.7(C-5),118.2(C-6),34.3(C-7),74.1(C-8),43.1(C-9),36.7(C-10),24.7(C-11),73.5(C-12),56.0(C-13),87.8(C-14),32.2(C-15),32.9(C-16),87.8(C-17),10.3(C-18),18.2(C-19),73.9(C-20),15.0(C-21),171.4(C-1′),21.7(C-2′),109.6(C-1″),152.2(C-2″),110.9(C-3″),134.8(C-4″),114.4(C-5″),131.4(C-6),167.2(C-7″),29.5(NCH3)。1H NMR(C5D5N,500MHz):δ3.89(1H,m,H-3),5.38(1H,br s,H-6),5.25(1H,dd,J=11.5,3.5Hz,H-12),2.05(3H,s,H-18),5.20(1H,q,J=6.5Hz,H-20),1.56(1H,d,J=6.0Hz,H-21),2.13(3H,s,H-2′),6.75(1H,d,J=8.5Hz,H-3″),7.42(1H,ddd,J=8.5,8.0,1.5Hz,H-4″),6.60(1H,t,J=7.0Hz,H-5″),8.37(1H,dd,J=8.0,2.0Hz,H-6″),2.81(3H,d,J=5.0Hz,NCH3)。经光谱数据分析与文献报道[Yoshikawa K,Okada N,Kann Y,Arihara S.Steroidal glycosides from the fresh stem of Stephanotis lutchuensis var.japonica(Asclepiadaceae).Chemical structures of stephanosides A–J.Chem Pharm Bull 1996;44:1790–1796.]的12-O-乙酰基-20-O-(N-甲基)邻氨基苯甲酰基肉珊瑚苷元完全一致。
实施例7:异开德苷元的制备和结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr1 16.7g,Fr2 13.0g,Fr3 14.5g。Fr1 16.7g经Rp-18柱层析,甲醇-水系统洗脱(50%→60%),TLC检测,合并相同部分,甲醇重结晶得到异开德苷元450mg。
异开德苷元,C28H42O8,无色针晶(甲醇)。ESI-MS(positive)m/z:529.2[M+Na]+。13C NMR(C5D5N,125MHz):38.9(C-1),32.0(C-2),71.5(C-3),43.2(C-4),139.7(C-5),118.6(C-6),34.7(C-7),74.1(C-8),43.9(C-9),37.3(C-10),25.6(C-11),74.5(C-12),56.7(C-13),88.8(C-14),33.6(C-15),33.4(C-16),87.5(C-17),10.9(C-18),18.1(C-19),74.8(C-20),15.1(C-21),171.2(C-1′),22.1(C-2′),167.0(C-1″),130.0(C-2″),136.9(C-3″),14.3(C-4″),12.5(C-5″)。1H NMR(C5D5N,500MHz):δ1.32(3H,s,H-19),2.01(3H,s,H-18),1.47(3H,s,H-21),1.60(3H,d,J=6.5Hz,H-4″),1.90(3H,s,H-5″),7.04(1H,qd,J=6.0,1.0Hz,H-2″)。经光谱数据分析与文献报道[Abe F,Okabe H,Yamauchi T,Honda K,Hayashi N.Pregnane glycosides from Marsdenia tomentosa.Chem Pharm Bull 1999;47:869–875.]的异开德苷元完全一致。
实施例8:开德苷元的制备与结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr1 16.7g,Fr2 13.0g,Fr3 14.5g。Fr1 16.7g经Rp-18柱层析,甲醇-水系统洗脱(50%→60%),TLC检测,合并相同部分,甲醇重结晶得到开德苷元210mg。
开德苷元,C28H42O8,无色针晶(甲醇)。ESI-MS(positive)m/z:529.2[M+Na]+。13C NMR(C5D5N,125MHz):39.3(C-1),32.3(C-2),71.8(C-3),43.6(C-4),140.4(C-5),118.9(C-6),35.1(C-7),74.6(C-8),44.3(C-9),37.4(C-10),25.5(C-11),75.2(C-12),56.6(C-13),89.0(C-14),33.7(C-15),34.0(C-16),87.9(C-17),11.4(C-18),18.5(C-19),74.8(C-20),15.7(C-21),167.5(C-1′),129.7(C-2′),138.0(C-3′),14.7(C-4′),12.5(C-5′),171.6(C-1″),22.4(C-2″)。1H NMR(C5D5N,500MHz):δ3.90(1H,m,H-3),5.39(1H,br s,H-6),5.18(1H,dd,J=11.5,4.0,H-12),2.24(3H,s,H-18),1.41(3H,s,H-19),5.08(1H,q,J=6.5Hz,H-20),1.49(3H,d,J=6.0Hz,H-21),6.56(1H,d,J=7.5Hz,H-3′),1.61(3H,d,J=7.0Hz,H-4′),1.92(3H,s,H-5′).2.02(1H,s,H-2″)。经光谱数据分析与文献报道[Tsukamoto S,Hayashi K,Mitsuhashi H.Studies on the constituents of Asclepiadaceae palnts.LX.Further studies on glycosides with a novel sugar chain containing a pair of optically isomeric sugars,D-and L-cymarose,from Cynanchum wilfordi.Chem Pharm Bull 1985;33:2294-2304.]的开德苷元一致。
实施例9:去乙酰开德苷元的制备与结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr116.7g,Fr213.0g,Fr314.5g。Fr213g经Rp-18柱层析,甲醇-水系统洗脱(50%→60%),经sephadex LH-20柱层析,甲醇洗脱,得到去乙酰开德苷元150mg。
去乙酰开德苷元C26H40O7,白色固体粉末。EI-MS(positive):m/z 487.3[M+Na]+.13C NMR(C5D5N,125MHz):39.1(C-1),32.0(C-2),71.5(C-3),43.4(C-4),140.0(C-5),118.8(C-6),35.0(C-7),74.3(C-8),44.1(C-9),37.2(C-10),25.6(C-11),74.3(C-12),57.0(C-13),88.8(C-14),34.2(C-15),32.9(C-16),88.6(C-17),11.6(C-18),18.3(C-19),70.8(C-20),19.4(C-21),167.7(C-1'),129.7(C-2'),137.9(C-3'),14.2(C-4'),12.3(C-5').1H NMR(C5D5N,500MHz):δ3.98(1H,m,H-3),5.34(1H,m,H-6),5.18(1H,dd,J=11.0,4.0Hz,H-12),2.06(3H,s,H-18),1.35(3H,s,H-19),4.42(1H,m,H-20),1.25(3H,d,J=5.5Hz,H-21),7.28(1H,dq,J=7.0,1.0Hz,H-3'),1.51(3H,dd,J=7.0,1.0Hz,H-4'),1.96(3H,s,H-5')。经光谱数据分析和理化性状测定与文献报道[Abe F,Okabe H,Yamacuchi T,Honda K,Hayashi N.Pregnane glycosides from Marsdenia tomenttosa Chem Pharm Bull 1999;47:869-875.]的去乙酰开德苷元完全一致。
实施例10:本波苷元的制备和结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr1 16.7g,Fr2 13.0g,Fr3 14.5g。Fr2 13.0g经Rp-18柱层析,甲醇-水系统洗脱(50%→60%),TLC检测,合并相同部分,甲醇重结晶得到本波苷元130mg。
本波苷元C30H40O7,amorphous powder.EI-MS(positive):m/z 535.3[M+Na]+.13C NMR(C5D5N,125MHz):39.1(C-1),32.0(C-2),71.6(C-3),43.4(C-4),140.0(C-5),118.9(C-6),35.0(C-7),74.2(C-8),44.2(C-9),37.2(C-10),25.7(C-11),74.8(C-12),56.9(C-13),88.8(C-14),34.2(C-15),32.9(C-16),88.6(C-17),11.7(C-18),18.3(C-19),70.9(C-20),19.3(C-21),165.9(C-1'),119.6(C-2'),145.2(C-3'),135.0(C-4'),128.6(C-5',C-9'),129.1(C-6',C-8'),130.5(C-7').1H NMR(C5D5N,500MHz):δ1.38(3H,s,H-19),1.92(3H,s,H-18),2.13(3H,s,H-21),3.85(1H,m,H-3),4.41(1H,m,H-20),5.27(1H,dd,J=11.5,4.5Hz,H-12),5.36(1H,br s,H-6),6.93(1H,d,J=16.0Hz,H-2′),7.50(2H,d,J=6.5Hz,H-5′,9′),7.22(2H,m,H-6′,8′),7.24(1H,m,H-7′),8.14(1H,d,J=16.0Hz,H-3′)。经光谱数据分析和理化性状测定与文献报道[Warashina T,Noro T.Steroidal glycosides from the root of Cynanchum caudatum M.Chem Pharm Bull1995;43:977-982]的本波苷元完全一致。
实施例11:加加明的制备和结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr1 16.7g,Fr2 13.0g,Fr3 14.5g。Fr1 16.7g经Rp-18柱层析,甲醇-水系统洗脱(50%→60%),TLC检测,合并相同部分,得到加加明320mg。
加加明C36H43NO8,amorphous powder.EI-MS(positive):m/z 618.3[M+H]+.13C NMR(C5D5N,125MHz):39.1(C-1),32.0(C-2),71.6(C-3),43.3(C-4),140.3(C-5),118.7(C-6),34.9(C-7),74.4(C-8),44.1(C-9),37.3(C-10),25.8(C-11),76.5(C-12),57.2(C-13),87.5(C-14),34.1(C-15),33.7(C-16),89.0(C-17),11.5(C-18),18.2(C-19),74.7(C-20),15.4(C-21),166.8(C-1'),120.3(C-2'),144.1(C-3'),136.0(C-4'),129.3(C-5',C-9'),128.6(C-6',C-8'),130.6(C-7'),151.4(C-1”),127.0(C-2”),137.5(C-3”),123.8(C-4”),153.8(C-5”),164.8(C-6”).1H NMR(C5D5N,500MHz):δ1.35(3H,s,H-19),1.56(3H,d,J=6.0Hz,H-21),2.11(3H,s,H-21),3.87(1H,m,H-3),5.31(1H,dd,J=11.0,4.0Hz,H-12),5.36(1H,br s,H-6),6.54(1H,d,J=16.0Hz,H-2′),7.35(2H,m,H-6′,8′),7.36(1H,m,H-7′),7.42(2H,d,J=5.5Hz,H-5′,9′),7.84(1H,d,J=16.0Hz,H-3′),7.21(1H,dd,J=7.5,4.5Hz,H-5″),8.32(1H,br d,J=7.5Hz,H-4″),8.83(1H,br d,J=4.5Hz,H-6″),9.52(1H,s,H-2″).经光谱数据分析和理化性状测定与文献报道[Tsukamoto S,Hayashi K,Mitsuhashi H.Studies on the constituents of Asclepiadaceae palnts.LX.Further studies on glycosides with a novel sugar chain containing a pair of optically isomeric sugars,D-and L-cymarose,from Cynanchum wilfordi.Chem Pharm Bull 1985;33:2294-2304.]的加加明完全一致。
实施例12:Kidjoranine的制备和结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr1 16.7g,Fr2 13.0g,Fr3 14.5g。Fr2 13g经Rp-18柱层析,甲醇-水系统洗脱(50%→60%),TLC检测,合并相同部分,经制备HPLC及Sephadex LH-20柱层析,得到Kidjoranine 480mg。
Kidjoranine,C30H38O7,amorphous powder.EI-MS(positive):m/z 533.3[M+Na]+.13C NMR(C5D5N,125MHz):39.1(C-1),32.0(C-2),70.5(C-3),43.3(C-4),140.3(C-5),118.4(C-6),34.7(C-7),74.3(C-8),44.5(C-9),37.3(C-10),25.0(C-11),73.6(C-12),58.1(C-13),92.4(C-14),34.0(C-15),33.0(C-16),89.5(C-17),10.6(C-18),18.3(C-19),209.8(C-20),27.6(C-21),165.8(C-1'),120.0(C-2'),144.9(C-3'),135.0(C-4'),128.5(C-5',C-9'),129.3(C-6',C-8'),130.5(C-7').1H NMR(C5D5N,500MHz):δ1.45(3H,s,H-19),2.08(3H,s,H-18),2.53(3H,s,H-21),3.92(1H,m,H-3),5.25(1H,dd,J=11.5,3.5Hz,H-12),5.37(1H,br s,H-6),6.87(1H,d,J=16.0Hz,H-2′),7.37(2H,m,H-6′,8′),7.38(1H,m,H-7′),7.67(2H,d,J=5.5Hz,H-5′,9′),8.05(1H,d,J=16.0Hz,H-3′)。经光谱数据分析和理化性状测定与文献报道[Tsukamoto S,Hayashi K,Mitsuhashi H.Studies on the constituents of Asclepiadaceae palnts.LX.Further studies on glycosides with a novel sugar chain containing a pair of optically isomeric sugars,D-and L-cymarose,from Cynanchum wilfordi.Chem Pharm Bull 1985;33:2294-2304.]的kidjoranine完全一致。
实施例13:12-O-惕各酰基-20-O-惕各酰基肉珊瑚苷元的制备和结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇溶液(即硫酸溶于甲醇形成硫酸甲醇溶液,其中硫酸甲醇溶液中硫酸的浓度为0.2mol/L)70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr1 16.7g,Fr2 13.0g,Fr3 14.5g。Fr2 13g经Rp-18(反向十八烷基烷化硅胶)柱层析,甲醇-水系统洗脱(甲醇-水系统中甲醇的体积百分比50%→60%),TLC(薄层色谱法)检测,合并相同部分,经制备HPLC及sephadex LH-20柱层析,得到化合物3(51mg)。
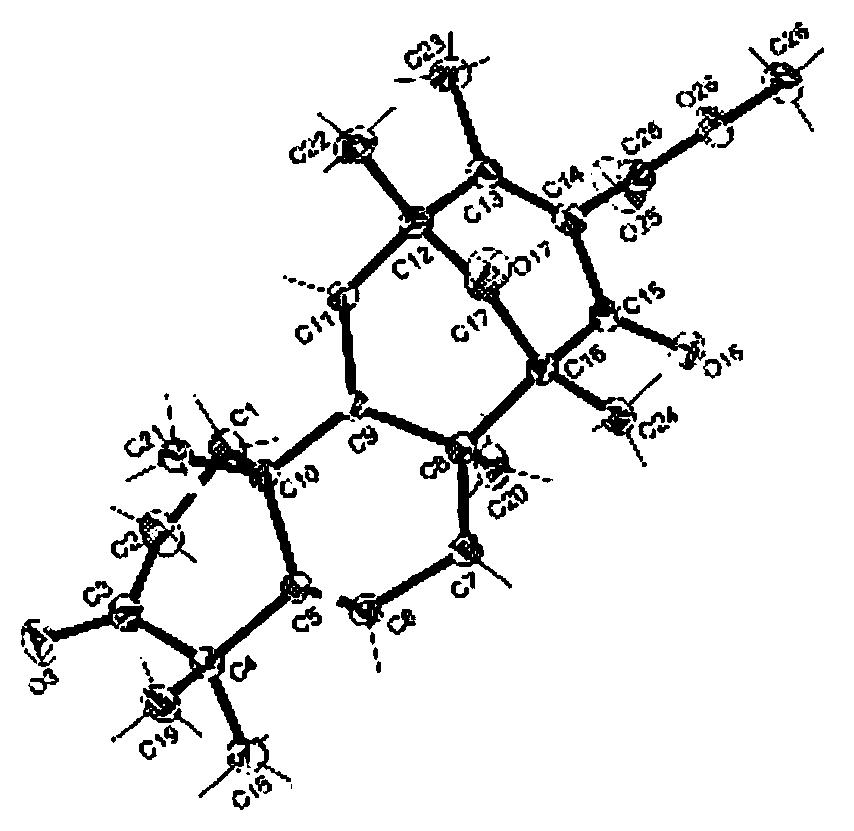
化合物3的结构鉴定白色无定型粉末,对Liebermann-Burchard反应呈阳性,表明其分子中存在甾核结构。高分辨质谱确定其分子式为C31H46O8(547.3273[M+H]+,计算值547.3271)。13C NMR和DEPT谱(125MHz,C5D5N)显示2含有31个碳原子,其中7个CH3、7个CH2、7个CH和10个季碳。化合物2的甾核部分的NMR谱数据与文献所描述的肉珊瑚苷元的数据相近[Li XY,Sun HX,Ye YP,Chen FY,Tu J,Pan YJ.Four new immunomodulating steroidal glycosides from the stems of Stephanotis mucronata.Steroids,2006,71:683-690.],只是化合物2的13C NMR,DEPT和1H NMR谱显示以下二组数据:(1)碳谱167.75(s),130.63(s),136.81(d),14.22(q),12.34(q),氢谱6.88(dq,J=7.0,1.0Hz),1.65(dd,J=7.0,1.0Hz),2.02(s);(2)碳谱166.60(s),129.57(s),135.89(d),14.03(q),12.12(q),氢谱6.80(dq,J=7.0,1.0Hz),1.54(dd,J=7.0,1.0Hz),1.78(s);显示分子中存在着两个惕各酰基。在化合物2的HMBC谱中,在δ167.75的季碳信号与甾体母核含氧的12位碳(δ74.72)上的氢[δ5.26(dd,J=11.0,4.0Hz)]相关,δ16,6.60的季碳信号与甾体母核含氧的20位碳(δ73.66)上的氢[δ5.04(q,J=6.0Hz)]相关,显示这两个惕各酰基分别连接在连接在甾体母核的12位与20位羟基上。因此化合物3的结构为12-O-惕各酰基-20-O-惕各酰基肉珊瑚苷元。
12-O-惕各酰基-20-O-惕各酰基肉珊瑚苷元的理化数据C31H46O8,白色无定形粉末,熔点136-138℃。EI-MS(positive):m/z 547.3[M+H]+.HR-EI-MS:547.3273([C31H46O8+H]+;calc.547.3271);569.3088([C31H46O8+Na]+;calc.569.3090).13C NMR(C5D5N,125MHz):38.9(C-1),32.0(C-2),71.5(C-3),43.3(C-4),140.1(C-5),118.7(C-6),34.9(C-7),74.3(C-8),43.9(C-9),37.1(C-10),25.7(C-11),74.7(C-12),56.9(C-13),88.8(C-14),34.1(C-15),33.8(C-16),87.7(C-17),11.4(C-18),18.2(C-19),73.7(C-20),15.3(C-21),167.8(C-1'),130.6(C-2'),136.7(C-3'),14.2(C-4'),12.3(C-5'),166.6(C-1”),129.6(C-2”),135.9(C-3”),14.3(C-4”),12.1(C-5”).1H NMR(C5D5N,500MHz):δ3.83(1H,dq,10.0,5.0Hz,H-3),5.36(1H,d,4.5Hz,H-6),5.26(1H,dd,11.0,4.0Hz,H-12),2.04(3H,s,H-18),1.35(3H,s,H-19),5.04(1H,q,6.0Hz,H-20),1.43(3H,d,6.0Hz,H-21),6.88(1H,dq,7.0,1.0Hz,H-3'),1.65(3H,dd,7.0,1.0Hz,H-4'),2.02(3H,s,H-5'),6.80(1H,dq,7.0,1.0Hz,H-3”),1.54(3H,dd,7.0,1.0Hz,H-4”),1.78(3H,s,H-5”).
实施例14:12-O-N-甲基邻氨基苯甲酰基去乙酰萝藦苷元(23)的制备和结构鉴定
黑鳗藤5kg粉碎,乙醇水溶液(含水的体积百分数为5%)渗漉,乙醇提取物用乙酸乙酯萃取,乙酸乙酯萃取物以0.2N硫酸甲醇溶液(即硫酸溶于甲醇形成硫酸甲醇溶液,其中硫酸甲醇溶液中硫酸的浓度为0.2mol/L)70℃水解5小时,碳酸氢钠中和,浓缩,浓缩物以乙酸乙酯萃取,萃取物即总苷元192g。总苷元192g上硅胶柱层析,以二氯甲烷、二氯甲烷-甲醇系统梯度洗脱,得到Fr1 16.7g,Fr2 13.0g,Fr3 14.5g。Fr2 13g经Rp-18柱层析,甲醇-水系统洗脱(50%→60%),TLC检测,合并相同部分,经制备HPLC及sephadex LH-20柱层析,得到化合物23 116mg。
化合物23的结构鉴定白色无定型粉末,其甲醇溶液显示强烈的蓝色荧光,显示可能有N-甲基邻氨基苯甲酰基基团的存在,对Liebermann-Burchard反应呈阳性,表明其分子中存在甾核结构。高分辨质谱确定其分子式为C29H39NO7(514.2803[M+H]+,计算值514.2805)。13C NMR和DEPT谱(125MHz,C5D5N)显示12含有29个碳原子,其中4个CH3、7个CH2、8个CH和10个季碳。化合物12的甾核部分的NMR谱数据与文献所描述的去乙酰萝藦苷元的数据相近[Ye YP,Li XY,Sun HX,Chen FY,Pan YJ.Immunomodulating Steroidal Glycosides from the Roots of Stephanotis mucronata.Helvetica Chimica Acta 2004;87:2378-2384.],只是化合物12的13C NMR,DEPT和1H NMR谱显示以下二组数据:(1)碳谱110.55(s),152.74(s),111.39(d),134.94(d),114.45(d),131.65(d),167.53(s),29.29(q),氢谱6.66(d,J=8.5Hz),7.42(ddd,J=8.5,8.0,1.5Hz),6.73(td,J=7.5,1.5Hz),8.16(dd,J=8.0,1.5Hz),8.21(q,J=5.0Hz),2.69(d,J=5.0Hz),显示分子中存在着N-甲基邻氨基苯甲酰基。在化合物12的HMBC谱中,在δ167.53的季碳信号与甾体母核含氧的12位碳(δ73.7)上的氢[δ5.31(dd,J=11.5,4.0Hz)]相关,显示N-甲基邻氨基苯甲酰基连接在甾体母核的12位羟基上。因此化合物23的结构为12-O-N-甲基邻氨基苯甲酰基去乙酰萝藦苷元。
12-O-N-甲基邻氨基苯甲酰基去乙酰萝藦苷元的理化数据:C29H39NO7,白色无定形粉末,熔点170-173℃。HR-ESI-MS:514.2803([C29H39NO7+H]+;计算值:514.2805);536.2620([C29H39NO7+Na]+;计算值:536.2624)。13C NMR(C5D5N,125MHz):39.1(C-1),32.0(C-2),71.5(C-3),43.3(C-4),140.3(C-5),118.4(C-6),34.7(C-7),74.3(C-8),44.4(C-9),37.3(C-10),25.1(C-11),73.1(C-12),58.3(C-13),92.4(C-14),33.8(C-15),33.2(C-16),89.6(C-17),10.9(C-18),18.3(C-19),209.9(C-20),27.7(C-21),110.55(C-1'),152.7(C-2'),111.4(C-3'),134.9(C-4'),114.5(C-5'),131.7(C-6'),167.5(C-7'),29.3(NCH3)。1H NMR(C5D5N,500MHz):δ3.90(1H,m,H-3),5.33(1H,d,4.5Hz,H-6),5.31(1H,dd,11.5,4.0Hz,H-12),2.07(3H,s,H-18),1.39(3H,s,H-19),2.39(3H,s,H-21),6.66(1H,d,8.5Hz,H-3'),7.42(3H,ddd,8.5,8.0,1.5Hz,H-4'),6.73(3H,td,7.5,1.5Hz,H-5'),8.16(1H,dd,8.0,1.5Hz,H-6'),2.69(3H,d,5.0Hz,H-NCH3),8.21(1H,q,4.5Hz,H-NH)。
实施例15:告达庭苷元3-硫酸酯的制备
告达庭苷元(1.7g,3.5mmol)抽真空氮气保护,加入吡啶4mL,三氧化硫-吡啶复合物(2.2g,14mmol);70℃,搅拌4h。后处理,浓缩至干,加入甲醇20mL,阳离子树脂10g,搅拌12h。过滤,浓缩,得黄色固体2.9g。分离纯化,NMR和MS测定结构,鉴定为告达亭苷元3-硫酸酯,其实验数据:13C NMR(C5D5N,125MHz):38.5(C-1),28.7(C-2),75.8(C-3),39.7(C-4),138.9(C-5),119.0(C-6),34.2(C-7),73.6(C-8),43.6(C-9),36.7(C-10),24.3(C-11),72.0(C-12),57.2(C-13),91.5(C-14),33.5(C-15),32.2(C-16),88.9(C-17),10.5(C-18),18.0(C-19),209.2(C-20),27.4(C-21),165.1(C-1′),113.6(C-2′),165.1(C-3′),37.6(C-4′),21.3(C-5′,9′),21.1(C-6′,8′),16.4(C-7′)。
实施例16:青阳参苷元3-硫酸酯的制备
青阳参苷元(200mg,0.4mmol)抽真空氮气保护,加入吡啶2mL,三氧化硫-吡啶复合物(127mg,0.8mmol);70℃,搅拌4h。后处理,浓缩至干,加入甲醇10mL,阳离子树脂5g,搅拌12h。过滤,浓缩,得黄色液体213mg。分离纯化,NMR和MS测定结构,鉴定为青阳参苷元3-硫酸酯,其实验数据:HR-ESI-MS:579.1906([C28H36O11S-H]-,计算值:579.1900)。13C NMR(C5D5N,125MHz):38.9(C-1),29.0(C-2),78.2(C-3),40.0(C-4),139.2(C-5),119.5(C-6),34.8(C-7),74.4(C-8),44.5(C-9),37.2(C-10),25.1(C-11),73.4(C-12),58.4(C-13),89.5(C-14),33.2(C-15),33.9(C-16),92.5(C-17),10.8(C-18),18.1(C-19),209.7(C-20),27.7(C-21),165.4(C-1′),122.1(C-2′),132.4(C-3′),116.2(C-4′),163.6(C-5′)。
实施例17
按实施例1的方法制得肉珊瑚苷元,按常规加注射用水,精滤,灌封灭菌制成注射液。
实施例18
按实施例1的方法制得肉珊瑚苷元,将其溶于无菌注射用水中,搅拌使溶,用无菌抽滤漏斗过滤,再无菌精滤,分装于安瓿瓶中,低温冷冻干燥后无菌熔封得粉针剂。
实施例19
按实施例1的方法制得肉珊瑚苷元,与赋形剂按一定比例混合,制成粉剂。
实施例20
按实施例1的方法制得肉珊瑚苷元,与赋形剂按一定比例混合,制粒压片。
实施例21
按实施例1的方法制得肉珊瑚苷元,按常规口服液制法制成口服液。
实施例22
按实施例1的方法制得肉珊瑚苷元,按其与赋形剂按一定比例混合,制成胶囊或颗粒剂或冲剂。
为了更好地理解本发明的优异性,以下将用本发明的式I化合物肉珊瑚苷元的药理作用结果即试验例子来说明,但并不以此来限定本发明。
试验例1.肉珊瑚苷元的抗抑郁活性
实验方法:采用药理实验方法学中的小鼠强迫游泳和小鼠悬尾实验(这些方法见徐叔云等.药理试验方法学.人民卫生出版社,2005:807-808中的描述)进行试验,均采用亚急性处理(24小时内2次给药),腹腔给药方式。实验设置溶剂对照,以氟西汀和丙咪嗪为阳性对照。
给药方式:肉珊瑚苷元设定0.05,0.1,0.25,0.5,1,5mg/kg共6个剂量组。实验采用雄性昆明小鼠,随机分组。测试前所有受试样品均腹腔给予,给药体积0.1ml/10g。给药频次及时间、测试时间为2次,分别在0和19h,在第1次给药后24h后测试。阳性对照氟西汀剂量15mg/kg,丙咪嗪剂量15mg/kg。
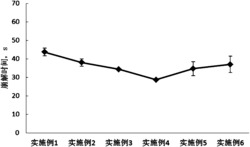
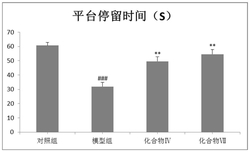
实验结果:与生理盐水对照相比,氟西汀(15mg/kg)和丙咪嗪(15mg/kg)显著缩短小鼠强迫游泳不动时间,表现出显著的抗抑郁活性;肉珊瑚苷元(0.05~0.5mg/kg)也具有显著的抗抑郁活性,且具有明显的剂量效应关系,其中最佳剂量为0.1mg/kg(见表2)。
表2肉珊瑚苷元在小鼠强迫游泳测试中的作用
注:*表示与生理盐水组相比有显著统计学差异。
与生理盐水对照相比,氟西汀和丙咪嗪显著缩短小鼠悬尾测试不动时间,肉珊瑚苷元未显著缩短小鼠悬尾测试的不动时间,但0.1、0.5和0.05mg/kg组表现出抗抑郁趋势,0.5mg/kg为最佳剂量(见表3)。
表3肉珊瑚苷元在小鼠悬尾测试中的作用
注:*表示与生理盐水组相比有显著统计学差异。
试验例2:肉珊瑚苷元的初步急性毒性试验
试验方法:取20只清洁级ICR雄性小鼠,随机分成两组,实验前禁食12h,按肉珊瑚苷元的最大溶解量和最大给药体积多次给药。其中,口服组采用超纯水配制,其最大溶解量为1.5mg/ml,24小时内分4次灌胃给药,每次0.3ml/10g体重,共计给药量为180mg/kg。腹腔注射组采用生理盐水配制,其最大溶解量为1.0mg/ml,24小时内分4次腹腔注射给药,每次0.3ml/10g体重,共计给药量为120mg/kg。给药后观察小鼠的毒性反应,连续观察7天。
试验结果:在最大溶解量和最大给药体积下,肉珊瑚苷元灌胃给药180mg/kg和腹腔注射给药120mg/kg,小鼠状态均良好,体重增长平稳,未见有明显毒副反应。
表1中的抗抑郁化合物3、抗抑郁化合物23以及R1为硫酸酯或醋酸酯,其他同抗抑郁化合物3、抗抑郁化合物23,也经过抗抑郁活性测试和初步急性毒性试验,表明上述化合物具有显著的抗抑郁活性,并且,未见有明显毒副反应。
一种抗抑郁化合物专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0