

IPC分类号 : C07C49/513,C07C45/80,C07C45/79,C07J73/00,C07C33/14,C07C29/86,C07C29/76,C07C29/78,A61K31/122,A61K31/047,A61K31/58,A61P5/30




专利摘要
结构式(I)所示的异海松烷型二萜衍生物(1‑7),含其为活性成分和药用载体所组成的药物组合物,该异海松烷型二萜衍生物(1–7)及其药物组合物的制备方法,其在制备治疗人类疾病的药物中的应用,特别是在制备治疗雌激素介导的各类疾病的药物中的应用,以及其用作为雌激素生物合成调节剂。
权利要求
1.结构式(I)所示的异海松烷型二萜衍生物2-3,
2.药物组合物,其中含有治疗有效量的权利要求1所述的式(I)所示的异海松烷型二萜衍生物2-3中的任一种和药学上可接受的载体。
3.权利要求1所述的式(I)所示的异海松烷型二萜衍生物2-3在制备雌激素生物合成调节剂中的应用。
4.权利要求1所述的式(I)所示的异海松烷型二萜衍生物2-3在制备治疗由雌激素介导的各类疾病的药物中的应用。
5.权利要求2所述的药物组合物在制备雌激素生物合成调节剂中的应用。
6.权利要求2所述的药物组合物在制备由雌激素介导的各类疾病的药物中的应用。
7.制备权利要求1所述的式(I)异海松烷型二萜衍生物2-3的方法,其特征在于取黄毛豆腐柴叶枝部分,干燥,粉碎,用80%乙醇在室温下提取3次,合并乙醇提液,过滤,减压浓缩;将该提取液悬浮于水溶液中,用乙酸乙酯萃取,乙酸乙酯萃取部分减压浓缩后,用氯仿溶解吸附于硅胶上,室温放置挥干溶剂,经硅胶柱层析,用1:0→0:1的氯仿/丙酮梯度洗脱,得到A-E五个部分;其中C部分用MCI脱色,95%甲醇/水洗脱,浓缩洗脱液后用RP-18反相硅胶再次分离,甲醇/水梯度洗脱,得到C1-C8馏分段;C4用Sephadex LH-20凝胶进一步分离,甲醇洗脱,得到C4.1-C4.5馏分段;C4.3部分再经过正相硅胶分离,石油醚/丙酮,8:1-1:1梯度洗脱,分成C4.3.1-C4.3.8馏分段,衍生物2从C4.3.5馏分中重结晶得到,馏分段C4.3.4经过HPLC,乙腈/水,48:52,3mL/min进一步的纯化得到衍生物3。
8.制备权利要求2所述的药物组合物的方法,其特征在于取黄毛豆腐柴叶枝部分,干燥,粉碎,用80%乙醇在室温下提取3次,合并乙醇提液,过滤,减压浓缩;将该提取液悬浮于水溶液中,用乙酸乙酯萃取,乙酸乙酯萃取部分减压浓缩后,用氯仿溶解吸附于硅胶上,室温放置挥干溶剂,经硅胶柱层析,用1:0→0:1的氯仿/丙酮梯度洗脱,得到A-E五个部分;其中C部分用MCI脱色,95%甲醇/水洗脱,浓缩洗脱液后用RP-18反相硅胶再次分离,甲醇/水梯度洗脱,得到C1-C8馏分段;C4用Sephadex LH-20凝胶进一步分离,甲醇洗脱,得到C4.1-C4.5馏分段;C4.3部分再经过正相硅胶分离,石油醚/丙酮,8:1-1:1梯度洗脱,分成C4.3.1-C4.3.8馏分段,衍生物2从C4.3.5馏分中重结晶得到,馏分段C4.3.4经过HPLC,乙腈/水,48:52,3mL/min进一步的纯化得到衍生物3;将上述步骤所获得的异海松烷型二萜衍生物2-3分别加入可药用载体得药物组合物。
说明书
技术领域:
本发明属于药物技术领域,具体地说,涉及一类异海松烷型二萜衍生物,含其为有效成分的治疗雌激素相关疾病的药物组合物,特别是在制备治疗由雌激素介导的各类疾病的药物中的应用,其制备方法,以及其用作为雌激素生物合成调节剂。
背景技术:
雌激素是人体内一种重要的内源性物质,介导了许多重要的生理功能。雌激素主要有三类,活性最高的是雌二醇(17β-estradiol,E2),其次是雌酮(estrone,E1),活性最低的为雌三醇(estriol,E3),其生物学效应主要通过与雌激素受体(estrogen receptor α/β)相结合,激活其转录或非转录活性,在生殖、免疫、骨骼、心血管和中枢神经统中发挥重要作用。雌激素缺乏常导致骨质疏松、冠心病、阿尔茨海默症、肥胖等常见疾病,而雌激素相对过量则是诸多肿瘤(如乳腺癌)发生、发展、转移等的直接原因。生物体内,胆固醇通过一系列酶促反应合成雌激素,其限速步骤是由芳香化酶催化雄激素底物转化为雌激素。芳香化酶在卵巢、脂肪组织及骨骼中均有表达,但在不同组织中使用不同启动子,其调控机制也不一样。
在我国,随着社会、经济的发展,近十年来,许多老年性疾病(如心脑血管疾病、骨质疏松症及乳腺癌)的发病率、死亡率和危险因素呈直线上升趋势。例如,由骨质疏松症导致的脊柱或胯骨骨折是老年人发病和死亡的一个主要原因。乳腺癌已经是患病率最高的妇女恶性肿瘤,严重影响绝经后妇女的生活质量。这些疾病的发生都与雌激素在机体内的合成代谢紊乱和/或其介导的信号途径失调有关,因此发现新的雌激素合成调节剂对于这些重大疾病的治疗具有重要意义。
天然产物由于其分子骨架及取代基的多样性,是天然的小分子物质结构数据库,也是现代药物研发的重要源泉。黄毛豆腐柴具有活血散淤、强筋健骨、驱风止痛的功能。实际上,黄毛豆腐柴的干燥茎就是一种壮药叫战骨,具有明显的抗炎、消肿和止痛,改善微循环、保护坐骨神经和软组织损伤作用。在民间常常被用于治疗肩周炎、骨质增生、肥大性脊椎炎、淋巴结炎、腰腿痛、肝区疼痛。然而,对其化学成分及生物活性的研究却鲜有报道。
迄今,现有技术中无此类异海松烷型二萜衍生物(式I所示1-7)的报道,也没有其作为有效成分的药物组合物的报道,也没有此类异海松烷二萜衍生物(式I所示1-9)及其药物组合物在制备雌激素生物合成调节剂和在制备治疗雌激素介导的疾病的药物中的应用的报道。
发明内容:
本发明的目的在于提供一类新的具有药用价值的式(I)所示的异海松烷二萜衍生物(1–7),含有能够调节雌激素生物合成并治疗雌激素介导的各类疾病有效量的异海松烷二萜衍生物(1–7)(I)及药用载体的作为雌激素合成调节剂并治疗雌激素介导的疾病的药物组合物,异海松烷二萜衍生物(1–7)及其药物组合物的制备方法,该类化合物或其药物组合物在制备雌激素生物合成调节剂和在制备治疗雌激素介导的各类疾病的药物中的应用。
为了实现本发明的上述目的,本发明提供了如下的技术方案:
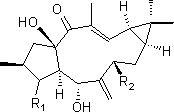
结构式(I)所示的异海松烷型二萜衍生物(1-7),
本发明同时提供了一种药物组合物,其中含有治疗有效量的权利要求1所述的式(I)所示的异海松烷型二萜衍生物(1-7)中的任一种和药学上可接受的载体。
本发明同时还提供了所述的式(I)所示的异海松烷型二萜衍生物(1-7)在制备雌激素生物合成调节剂中的应用。
所述的式(I)所示的异海松烷型二萜衍生物(1-7)在制备治疗由雌激素介导的各类疾病的药物中的应用。
所述的式(I)所示的异海松烷型二萜衍生物(1-7)用作为雌激素生物合成调节剂。
所述的药物组合物在制备雌激素生物合成调节剂中的应用。
所述的药物组合物在制备由雌激素介导的各类疾病的药物中的应用。
所述的药物组合物用作为雌激素生物合成调节剂。
本发明此外还提供了制备式(I)异海松烷型二萜衍生物(1-7)的方法,取黄毛豆腐柴叶枝部分,干燥,粉碎,用80%乙醇在室温下提取3次,合并乙醇提液,过滤,减压浓缩;将该提取液悬浮于水溶液中,用乙酸乙酯萃取,乙酸乙酯萃取部分减压浓缩后,用氯仿溶解吸附于硅胶上,室温放置挥干溶剂,经硅胶柱层析,用1:0→0:1的氯仿/丙酮梯度洗脱,得到A-E五个部分;其中C部分用MCI脱色,95%甲醇/水洗脱,浓缩洗脱液后用RP-18反相硅胶再次分离,甲醇/水梯度洗脱,得到C1-C8馏分段;C4用Sephadex LH-20凝胶进一步分离,甲醇等度洗脱,得到C4.1-C4.5馏分段;C4.3部分再经过正相硅胶分离,石油醚/丙酮,8:1-1:1梯度洗脱,分成C4.3.1-C4.3.8馏分段,化合物2和7分别从C4.3.5和C4.3.7两个馏分中重结晶得到,馏分段C4.3.4经过HPLC,乙腈/水,48:52,3mL/min进一步的纯化得到化合物3和4,C4.4馏分经过HPLC,乙腈/水,46:54,3mL/min分离纯化得到化合物1,5和6。
制备含异海松烷型二萜衍生物(1-7)的药物组合物的方法是以异海松型二萜衍生物(1-7)为原料,加入可药用载体或赋形剂。
本发明化合物用作药物时,可以直接使用,或者以药物组合物的形式使用。该药物组合物含有0.1–99%,优选为0.5–90%的本发明化合物,其余为药物学上可接受的,对人和动物无毒和惰性的可药用载体和/或赋形剂。
所述的药用载体或赋形剂是一种或多种固体、半固体和液体稀释剂、填料以及药物制品辅剂。将本发明的药物组合物以单位体重服用量的形式使用。本发明的药物可经注射(静注、肌注)和口服两种形式给药。
附图说明:
图1.化合物2抑制雌激素生物合成作用。
图2.化合物3促进雌激素生物合成作用。
图3.化合物2抑制雌激素合成效果与时间关系。
图4.化合物3促进雌激素合成效果与时间关系。
图5.化合物2抑制芳香化酶mRNA生成。
图6.化合物3促进芳香化酶mRNA生成。
图7.化合物2抑制芳香化酶生成。
图8.化合物3促进芳香化酶生成。
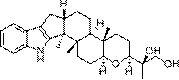
图9为本发明的式(I)异海松烷型二萜衍生物(1-7)的结构示意图。
具体实施方式:
为了更好地理解本发明的实质,下面将结合附图,用本发明的试验例来说明本发明式(I)异海松烷型二萜衍生物(1-7)的药理作用结果,但不以此试验例来限定本发明。
试验例1:
式(I)异海松烷型二萜衍生物(1-7)对雌激素体内生物合成调节作用:
1试验方法
1.1基于细胞的雌激素生物合成试验。将KGN细胞细胞接种于24孔板中过夜。第二天,用无血清的DMEM/F-12培养基代替培养基,预处理细胞24小时,然后将睾酮(10nm)加入到每个孔中,再将细胞培养24小时。用磁微粒分离酶联免疫法(ELISA试剂盒,倍爱康生物科技,北京,中国)检测KGN细胞17β-雌二醇的含量。通过归一化到总的细胞蛋白质含量,检测结果可表示为对照的百分比。蛋白质检测用BCA蛋白检测试剂盒(Pierce,罗克福德,IL,美国)进行测定。
1.2实时定量PCR。根据制造商的方案,使用TRIzol试剂(Invitrogen)分离总细胞RNA。使用寡核苷酸(dT)18引物和SuperScript III逆转录酶(Invitrogen)逆转录总RNA。根据制造商的方案(Fermentas,Thermo Scientific),用荧光染料SYBR Green I对相同数量的互补DNA进行实时定量PCR。下面的引物对用于芳香化酶、启动子II、启动子II3和GAPDH:
5′-ACCCTTCTGCGTCGTGTC-3′/5′-TCTGTGGAAATCCTGCGTCTT-3′(aromatase sense/antisense),5′-TCCCTTTGATTTCCACAGGACTC-3′/5′-ATGCAGTAGCCAG GACCTGGT-3′(promoter II sense/antisense);5′-CACTCTACCCACTCAAGGGCA-3′/5′-TTGGCTTGAATTGCAGCATTT-3′(promoter I.3 sense/antisense);
5′-TGCACCACCAACTGCTTAGC-3′/5′-GGCATGGACTGTGGTCATGAG-3′(GAP-DH sense/antisense)。
芳香化酶、启动子II和启动子I.3的mRNA的含量在同一样品中归一化为内源性参考物(GAPDH)mRNA量。
1.3蛋白质印迹。细胞在加了蛋白酶抑制剂(Sigma-Aldrich)的RIPA缓冲液(Byotime,Haimen,China)中裂解。将细胞裂解物(50μg)进行10%SDS-PAGE电泳,转移到硝酸纤维素膜上(Bio-Rad,Hercules,CA,USA)。芳香化酶抗体(Epitomics,Burlingame,CA,USA),GAPDH抗体(Abgent,Suzhou,China),phospho-CREB抗体,CREB抗体,phospho-ERK抗体,phospho-JNK抗体(Cell Signaling Technology,Danvers,MA,USA),phospho-p38抗体,phospho-AKT抗体(Signaling Antibody LLC,College Park,MD,USA)。结合了辣根过氧化物酶(Pierce)的二抗用于蛋白质检测。使用增强的化学发光检测(Amersham Bioscience,Piscataway,NJ,USA)开发了膜。通过使用BCA蛋白检测试剂盒(Pierce)测定蛋白质浓度。
2.试验结果:
2.1通过雌激素生物合成试验(结果如图1-4所示),发现化合物2具有显著抑制雌激素生物合成的作用,IC50值为10.68±0.215μM;而化合物3表现出相反的(即促进雌激素生物合成)的效果,EC50值为15.49±0.148μM。而且这两个化合物对雌激素生物合成的影响具有呈现时间和计量的依赖性。
2.2通过实时定量PCR试验(结果如图5和6所示),发现经过化合物2和3处理过的KGN细胞中的芳香化酶mRNA的表达量显著的下降和增加,且呈现出剂量依赖关系。而检测芳香化酶的蛋白质印迹试验结果与定量PCR试验结果一致(结果如图7和8所示),经过化合物2和3处理过的KGN细胞中的芳香化酶蛋白的量明显降低和增加,且呈现出剂量依赖关系。
3、结论:
实验结果表明,化合物2具有显著抑制雌激素生物合成的作用,IC50值为10.68±0.215μM;而化合物3表现出促进雌激素生物合成的作用,EC50值为15.49±0.148μM。且这两个化合物都通过调节芳香化酶mRNA的表达来达到影响雌激素的生物合成的效果。
以下通过本发明的实施例进一步来阐明本发明的制备方法及药物组成,但并不以此来限定本发明。
实施例1:
异海松烷型二萜衍生物(1–7)(I)的制备:
异海松烷型二萜衍生物(1–7)(I)的提取分离:
实验中使用的半制备高效液相为Agilent 1260和Zorbax SB-C18(9.4mm×25cm)色谱柱;薄层色谱硅胶、柱层析硅胶(100-200目和200-300目)购自青岛美高集团有限公司;反向C18硅胶为Lichroprep RP-18gel(40-63μm,Merck,Darmstadt,Germany),葡聚糖凝胶为Sephadex LH-20(Pharmacia)。
采集黄毛豆腐柴(Premnafulva)叶枝部分,干燥,粉碎,用80%乙醇在室温下提取3次,合并乙醇提液,过滤,减压浓缩;将该提取液悬浮于水溶液中,用乙酸乙酯萃取,乙酸乙酯萃取部分减压浓缩后,用氯仿溶解吸附于硅胶上,室温放置挥干溶剂,经硅胶柱层析,用1:0→0:1的氯仿/丙酮梯度洗脱,得到A-E五个部分;其中C部分用MCI脱色(95%甲醇/水洗脱),浓缩洗脱液后用RP-18反相硅胶再次分离(甲醇/水梯度洗脱),得到C1-C8馏分段;C4用Sephadex LH-20凝胶进一步分离(甲醇等度洗脱),得到C4.1-C4.5馏分段;C4.3部分再经过正相硅胶分离(石油醚/丙酮,8:1-1:1梯度洗脱)分成C4.3.1-C4.3.8馏分段。化合物2和7分别从C4.3.5和C4.3.7两个馏分中重结晶得到。馏分段C4.4.4经过HPLC(乙腈/水,48:52,3mL/min)进行进一步的纯化得到化合物3和4。C4.4馏分经过HPLC(乙腈/水,46:54,3mL/min)分离纯化得到化合物1,5和6。以上化合物的结构通过
异海松烷型二萜衍生物(1–9)的结构数据:
旋光由SEPA-300和Jascomodel 1020旋光仪(Horiba,Tokyo,Japan)测定;红外光谱(IR)采用KBr压片法,由Tenor 27型红外光谱仪;紫外光谱由UV-2401A型紫外光谱仪(Shimadzu)测定;核磁共振谱(NMR)用Brucker AM-400型和DRX-500型超导核磁共振仪测定,以丙酮作为溶剂,TMS(四甲基硅烷)作内标;高分辨质谱(HREI-MS)用API Qstar Pulsar质谱仪测定。化合物的NMR数据如表1和表2所示。
表1异海松烷型二萜衍生物(1-7)(I)的
表2异海松型二萜衍生物(1-7)(I)的
化合物1
分子式:C20H32O4
分子量:336.23
性状:无色结晶
旋光
IR(KBr)vmax:3426,2934,1693,1452,1380,1230,1143,1040,907,543cm
UV/Vis(MeOH)λmax(logε):206(3.53)nm。
ESIMS[M+Na]
HRESIMS[M+Na]
化合物2
分子式:C20H32O2
分子量:304.24
性状:无色结晶
旋光
IR(KBr)vmax:3425,2931,1631,1381,1015,572cm
UV/Vis(MeOH)λmax(logε):208(3.81)nm。
ESIMS[M+Na]
HRESIMS[M+Na]
化合物3
分子式:C20H30O3
分子量:318.22
性状:无色结晶
旋光
IR(KBr)vmax:3422,2927,1649,1381,1268,1191,1026,615cm
UV/Vis(MeOH)λmax(logε):203(3.66),270(3.73)nm。
ESIMS[M+Na]
HRESIMS[M+Na]
化合物4
分子式:C20H30O3
分子量:318.22
性状:无色结晶
旋光
IR(KBr)vmax:3421,2928,2861,1650,1461,1383,1207,1037,619cm
UV/Vis(MeOH)λmax(logε):201(3.26),268(3.70)nm。
EIMS[M-H]
HRESIMS[M+Na]
化合物5
分子式:C20H32O3
分子量:320.24
性状:无色结晶
旋光
IR(KBr)vmax:3421,2929,1629,1459,1381,1040,635cm
UV/Vis(MeOH)λmax(logε):205(3.83)nm。
EIMS[M+Na]
HRESIMS[M+Na]
化合物6
分子式:C20H32O3
分子量:320.24
性状:无色结晶
旋光
IR(KBr)vmax:3423,2925,1630,1464,1382,1050cm
UV/Vis(MeOH)λmax(logε):203(3.57)nm。
EIMS[M+Na]
HRESIMS[M+Na]
化合物7
分子式:C20H34O3
分子量:322.25
性状:无色结晶
旋光
IR(KBr)vmax:3374,2929,1631,1384,1069,878,699cm
UV/Vis(MeOH)λmax(logε):206(3.92)nm。
EIMS[M-H]
HRESIMS[M+Na]
实施例2:
按实施例1的方法先制得异海松烷型二萜衍生物(1-7),分别用少量的DMSO溶解后,按常规加注射用水,精滤,灌封灭菌制成注射液。
实施例3:
按实施例1的方法先制得异海松烷型二萜衍生物(1-7),分别用少量的DMSO溶解后,将其溶于无菌注射用水中,搅拌使溶解,用无菌抽滤漏斗过滤,再无菌精滤,分装于安瓿中,低温冷冻干燥后无菌熔封得粉针剂。
实施例4:
将所分离得到的异海松烷型二萜衍生物(1-7),分别按其与赋形剂重量比为9:1的比例加入赋形剂,制成粉剂。
实施例5:
按实施例1的方法先制得异海松烷型二萜衍生物(1-7),分别按其与赋形剂重量比为5:1的比例加入赋形剂,制粒压片。
实施例6:
按实施例1的方法先制得异海松烷型二萜衍生物(1-7),分别按常规口服液制法制成口服液。
实施例7:
按实施例1的方法先制得异海松烷型二萜衍生物(1-7),分别按其与赋形剂重量比为5:1的比例加入赋形剂,制成胶囊。
实施例8:
按实施例1的方法先制得异海松烷型二萜衍生物(1-7),分别按其与赋形剂重量比为3:1的比例加入赋形剂,制成胶囊。
一类异海松烷型二萜衍生物,其药物组合物及其用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。












动态评分
0.0