










专利摘要
本发明提供了一种分离与分析石油组分中非碱性氮化合物的方法。该方法包括以下步骤:将石油组分溶解在有机溶剂中,加入烷基化试剂在0-80℃反应12-48h,过滤取滤液;将滤液与C5-C7的烷烃溶剂混合,分离出沉淀;将沉淀溶解在有机溶剂中,加入脱烷基试剂在50-150℃反应12-48h;使得到的混合液经过柱层析分离,除去含硫化合物,得到甲基化的非碱性氮化合物;采用气相色谱-质谱仪对得到的氮化物进行检测。本发明所提供的分离与分析石油组分中非碱性氮化合物的方法,能够将非碱性氮化物转化为N-甲基衍生物并实现其与烃类及其他杂原子化合物的复杂基质中完全分离,适用于石油组分中低含量非碱性氮化合物的分子组成分析。
权利要求
1.一种分离与分析石油组分中非碱性氮化合物的方法,其包括以下步骤:
步骤一:将石油组分溶解在有机溶剂中,然后加入烷基化试剂在0-80℃进行烷基化反应12-48h,之后进行过滤,取滤液;
步骤二:将所述滤液与C5-C7的烷烃溶剂混合,然后分离出沉淀;
步骤三:将所述沉淀溶解在有机溶剂中,然后加入脱烷基试剂在50-150℃进行脱烷基反应12-48h;
步骤四:使步骤三反应得到的混合液经过柱层析分离,先用非极性溶剂脱除含硫化合物,再用极性溶剂冲洗出甲基化的非碱性氮化合物;
步骤五:采用气相色谱-质谱联用仪对步骤四得到的甲基化的非碱性氮化合物进行检测,以得到石油组分中非碱性氮化合物的分子组成信息。
2.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,所述石油组分包括轻质原油、柴油馏分和减压瓦斯油馏分中的一种或几种的组合,所述石油组分的总氮含量为10-20000μg/g。
3.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,在步骤一中,所述有机溶剂包括乙醇、二氯甲烷和二氯乙烷中的一种或几种的组合,所述有机溶剂的用量为所述石油组分体积的1-20倍,优选为5-10倍。
4.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,在步骤一中,所述烷基化试剂包括硝酸银、四氟硼酸银、碘甲烷和溴乙烷中的一种或几种的组合,所述烷基化试剂的用量为所述石油组分中总氮元素摩尔量的10-200倍,优选为20-100倍。
5.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,在步骤二中,所述C5-C7的烷烃溶剂包括正戊烷、正己烷和正庚烷中的一种或几种的组合,所述C5-C7的烷烃溶剂的用量为所述石油组分体积的20-500倍,优选为100-300倍。
6.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,在步骤三中,所述有机溶剂包括二氯甲烷、三氯甲烷和乙腈中的一种或几种的组合,所述有机溶剂的用量为所述石油组分体积的1-20倍,优选为5-10倍。
7.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,在步骤三中,所述脱烷基试剂包括吡啶、4-二甲氨基吡啶和7-氮杂吲哚中的一种或几种的组合,所述脱烷基试剂的用量为所述石油组分中总氮元素摩尔量的10-100倍,优选为10-30倍。
8.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,在步骤四中,分离纯化所用的固定相为硅胶,所述硅胶的用量为所述石油组分重量的4-20倍;优选地,所述硅胶为能通过200-300目网筛的硅胶。
9.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,在步骤四中,脱除含硫化合物所用的非极性溶剂包括C5-C7的烷烃,所述C5-C7的烷烃的用量为所述石油组分体积的10-200倍,优选为20-80倍。
10.根据权利要求1所述的分离与分析石油组分中非碱性氮化合物的方法,其中,在步骤四中,冲洗出甲基化的非碱性氮化合物所用的极性溶剂包括二氯甲烷和/或三氯甲烷,所述二氯甲烷和/或三氯甲烷的用量为所述石油组分体积的5-50倍,优选为10-20倍。
说明书
技术领域
本发明涉及一种分离与分析石油组分中非碱性氮化合物的方法,属于石油分析技术领域。
背景技术
氮是石油组分中最重要的杂原子之一,其中的非碱性氮化物分子组成可以用于石油勘探研究油气运移方向,非碱性氮化合物对石化燃料的运输与存储的安定性产生不利影响,在石化燃料的使用过程中释放氮氧化物(NOx)等有害气体。因此,研究石油中非碱性氮化物组成是石油工业界关注的课题,然而非碱性氮化合物在石油中的浓度很低,需要将这些化合物从十分复杂的石油基质中分离出来才能进行有效分析。
非碱性氮化合物的分离与检测是石油化学研究的一个难题。目前对石油组分中的非碱性氮化合物主要采用开口柱色谱进行分离,配合不同配比的氧化铝和硅胶作为固定相,以不同配比的正己烷、三氯甲烷和甲醇作为流动相。由于石油是由包括烃类、含硫化合物、碱性氮化合物、非碱性氮化合物等各类化合物组成的连续分布的复杂混合体,单纯柱色谱很难分离得到高纯的非碱性氮化合物。并且单纯柱色谱方法经验性强,对操作技巧要求较高,因此对轻重不同的石油组分适用性较差,易造成非碱性氮化合物的丢失,对定性定量检测造成极大困扰。
深入研究石油中非碱性氮化合物的组成以及其在石油加工过程中的反应性能,需要一种实现非碱性氮化合物与烃类及其他杂原子化合物的完全分离,进而进行检测的技术。
发明内容
为了解决上述技术问题,本发明的目的在于提供一种分离与分析石油组分中非碱性氮化合物的方法,该方法能够对石油馏分中非碱性氮化合物分子组成进行详细表征。
为了达到上述目的,本发明提供了一种分离与分析石油组分中非碱性氮化合物的方法,其包括以下步骤:
步骤一:将石油组分溶解在有机溶剂中,然后加入烷基化试剂在0-80℃进行烷基化反应12-48h,之后进行过滤,取滤液;
步骤二:将所述滤液与C5-C7的烷烃溶剂混合,然后分离出沉淀;
步骤三:将所述沉淀溶解在有机溶剂中,然后加入脱烷基试剂在50-150℃进行脱烷基反应12-48h;
步骤四:使步骤三反应得到的混合液经过柱层析分离,先用非极性溶剂脱除含硫化合物,再用极性溶剂冲洗出甲基化的非碱性氮化合物(该甲基化的非碱性氮化合物为石油组分中原始非碱性氮化合物的氮原子上连接一个甲基的非碱性氮化合物甲基衍生物);
步骤五:采用气相色谱-质谱联用仪对得到的甲基化的非碱性氮化合物进行检测,以得到石油组分中非碱性氮化合物的分子组成信息。
在上述的方法中,采用气相色谱-质谱联用仪对甲基化的非碱性氮化合物进行检测的方法为本领域的常规方法,其中的气相色谱分析条件和质谱分析条件都是本领域技术人员能够根据本领域公知常识通过常规技术手段可以进行调节的,此处不再赘述。
在本发明所提供的分离与分析石油组分中非碱性氮化合物的方法中,优选地,所述石油组分包括轻质原油、柴油馏分和减压瓦斯油馏分中的一种或几种的组合。
在本发明所提供的分离与分析石油组分中非碱性氮化合物的方法中,优选地,所述石油组分的总氮含量为10-20000μg/g。
根据本发明的具体实施方案,优选地,在上述方法的步骤一中,所述有机溶剂包括乙醇、二氯甲烷和二氯乙烷等中的一种或几种的组合,所述有机溶剂的用量为所述石油组分体积的1-20倍,更优选为5-10倍。
根据本发明的具体实施方案,优选地,在上述方法的步骤一中,所述烷基化试剂包括硝酸银、四氟硼酸银、碘甲烷和溴乙烷等中的一种或几种的组合,所述烷基化试剂的用量为所述石油组分中总氮元素摩尔量的10-200倍,更优选为20-100倍。
根据本发明的具体实施方案,优选地,在上述方法的步骤二中,所述滤液与C5-C7的烷烃溶剂混合之前,可以先将滤液中的有机溶剂以及烷基化试剂去除,例如通过挥发等方法使其去除。
根据本发明的具体实施方案,优选地,在上述方法的步骤二中,所述C5-C7的烷烃溶剂包括正戊烷、正己烷和正庚烷等中的一种或几种的组合,所述C5-C7的烷烃溶剂的用量为所述石油组分体积的20-500倍,更优选为100-300倍。
根据本发明的具体实施方案,优选地,在上述方法的步骤二中,将所述滤液与C5-C7的烷烃溶剂混合后,可以超声1-3分钟,然后离心分离沉淀,并且可以将得到的沉淀重复离心2-3次。
根据本发明的具体实施方案,优选地,在上述方法的步骤三中,所述有机溶剂包括二氯甲烷、三氯甲烷和乙腈等中的一种或几种的组合,所述有机溶剂的用量为所述石油组分体积的1-20倍,更优选为5-10倍。
根据本发明的具体实施方案,优选地,在上述方法的步骤三中,所述脱烷基试剂包括吡啶、4-二甲氨基吡啶和7-氮杂吲哚等中的一种或几种的组合,所述脱烷基试剂的用量为所述石油组分中总氮元素摩尔量的10-100倍,更优选为10-30倍。
根据本发明的具体实施方案,优选地,在上述方法的步骤四中,使步骤三反应得到的混合液经洗涤和有机相浓缩后,再经过柱层析分离纯化,其中洗涤可以采用盐酸等,该盐酸的浓度和用量可以由本领域技术人员进行常规的调节。
根据本发明的具体实施方案,优选地,在上述方法的步骤四中,分离纯化所用的固定相为硅胶,所述硅胶的用量为所述石油组分重量的4-20倍;更优选地,所述硅胶的用量为所述石油组分重量的10倍。
根据本发明的具体实施方案,优选地,在上述方法的步骤四中,所述硅胶为能通过200-300目网筛的硅胶。
根据本发明的具体实施方案,优选地,在上述方法的步骤四中,脱除含硫化合物所用的非极性溶剂包括C5-C7的烷烃,所述C5-C7的烷烃的用量为所述石油组分体积的10-200倍,优选为20-80倍。
根据本发明的具体实施方案,优选地,在上述方法的步骤四中,冲洗出甲基化的非碱性氮化合物所用的极性溶剂包括二氯甲烷和/或三氯甲烷,所述二氯甲烷和/或三氯甲烷的用量为所述石油组分体积的5-50倍,优选为10-20倍。
在本发明上述方法的步骤四中,在采用C5-C7的烷烃进行洗脱以分离出含硫化合物后,再采用二氯甲烷和/或三氯甲烷进行洗脱以得到目标甲基化的非碱性氮化合物。
本发明所提供的分离与分析石油组分中非碱性氮化合物的方法能够将非碱性氮化合物转化为N-甲基衍生物,并实现其与烃类及其他杂原子化合物的完全分离,使微量非碱性氮化物可以利用气相色谱-质谱检测。本发明所提供的分离与分析石油组分中非碱性氮化合物的方法适用于石油组分中低含量非碱性氮化合物的分子组成分析,例如轻质原油、柴油馏分、减压瓦斯油馏分等,而且操作条件温和,能够完整、高效地分离出高纯度的甲基化的非碱性氮化合物,对其组成进行检测,能够反映出石油组分中原始非碱性氮化合物的分子组成信息。
附图说明
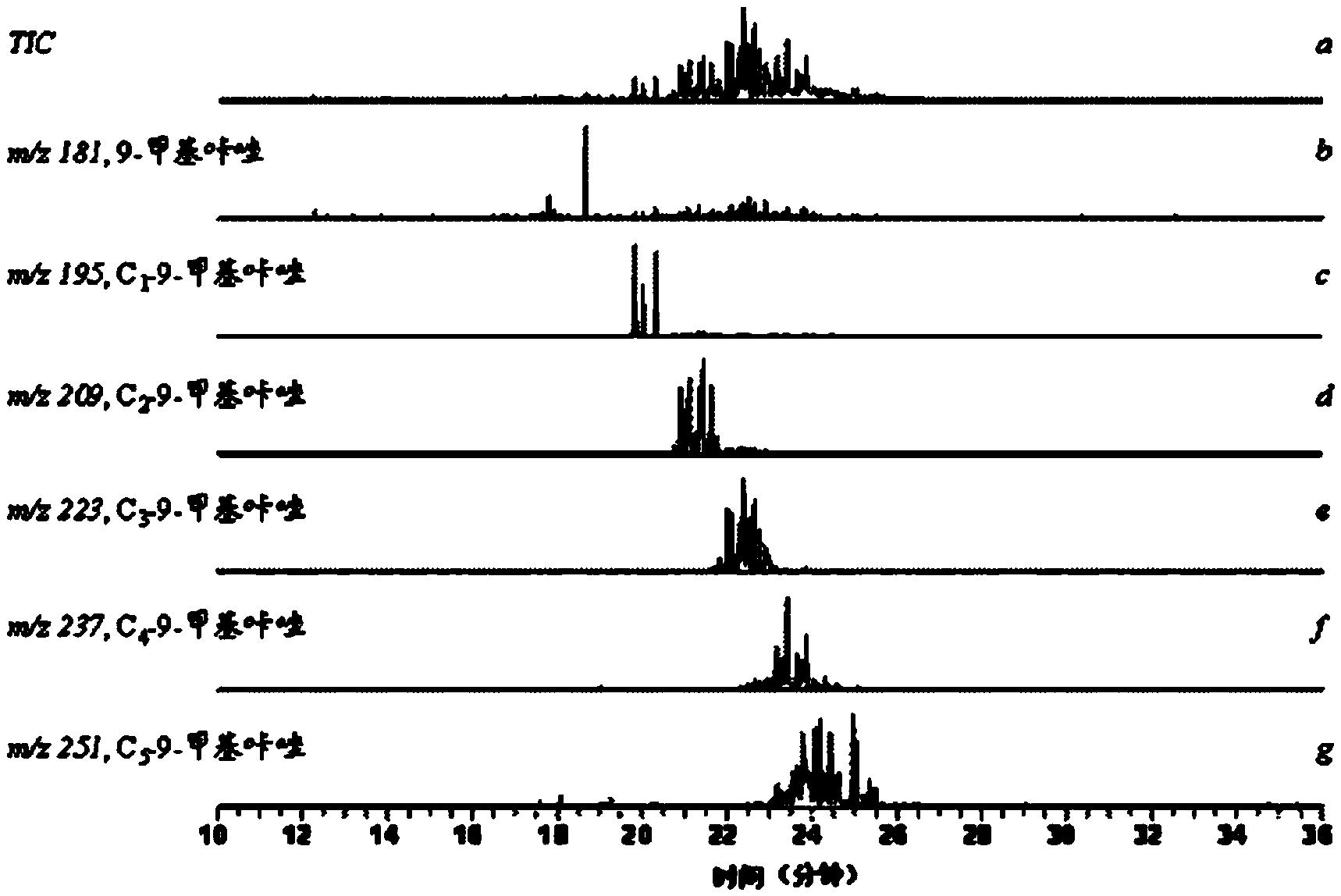
图1为实施例1中从柴油组分中分离其得到的非碱性氮化合物甲基衍生物的色谱图,其中,曲线a为通过GC-MS检测的非碱性氮化合物甲基衍生物的总离流色谱图;曲线b为通过GC-MS检测的质核比为181的质量色谱图,对应N-甲基咔唑化合物;曲线c为通过GC-MS检测的质核比为195的质量色谱图,对应C1-N-甲基咔唑化合物;曲线d为通过GC-MS检测的质核比为209的质量色谱图,对应C2-N-甲基咔唑化合物;曲线e为通过GC-MS检测的质核比为223的质量色谱图,对应C3-N-甲基咔唑化合物;曲线f为通过GC-MS检测的质核比为237的质量色谱图,对应C4-N-甲基咔唑化合物;曲线g为通过GC-MS检测的质核比为251的质量色谱图,对应C5-N-甲基咔唑化合物。
具体实施方式
为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现对本发明的技术方案进行以下详细说明,但不能理解为对本发明的可实施范围的限定。
实施例1
本实施例提供了一种分离与分析石油组分中非碱性氮化合物的方法,其具体步骤如下:
首先,将10克总氮含量为169μg/g的加氢精制柴油和50毫升二氯甲烷加入到250毫升的茄形瓶中,随后加入0.5克四氟硼酸银,在强烈搅拌下用注射器加入0.3毫升碘甲烷,将密封的茄形瓶在避光条件下在25℃下搅拌反应24小时,再加入0.2毫升碘甲烷,同样条件下再反应24小时。
然后,将反应生成的碘化银过滤后得到的滤液在氮气气氛中挥发除去二氯甲烷和过量碘甲烷,加入100毫升正己烷后,超声1分钟,然后离心沉淀其中的不溶部分,收集上清液;重复上述分离步骤5次,合并上清液。
最后,向离心得到的沉淀中加入10毫升三氯甲烷和0.5克4-二甲氨基吡啶在搅拌下加热至60℃回流反应24小时,所得反应液用10毫升浓度为2摩尔/升的盐酸洗涤,有机相通过旋转蒸发器浓缩后经过硅胶柱层析分离纯化,先使用60毫升正已烷洗脱含硫化合物,再使用50毫升二氯甲烷进行洗脱得到甲基化的非碱性氮化合物(即非碱性氮化合物甲基衍生物),其中硅胶为能通过200-300目网筛的硅胶,其用量为5克。
采用气相色谱-质谱联用仪对得到的甲基化的非碱性氮化合物进行检测,气相色谱分析采用美国Thermo公司FinniganTrace DSQ气相色谱-质谱联用仪,气相色谱条件:HP5-MS(30m×0.25mm×0.25μm)弹性石英管毛细柱。进样口温度为280℃,升温程序:40℃保持1分钟,10℃/min升至300℃,保持10分钟。质谱条件:EI电离源,电子能量70eV,灯丝电流300μA,倍增器电压1100V,离子源温度250℃,全扫描质量范围35-500amu,扫描周期1.5秒。结果如图1所示。
图1为实施例1中从柴油组分中分离其得到的非碱性氮化合物甲基衍生物(N-甲基取代咔唑类化合物)的色谱图,其中,曲线a为通过GC-MS检测的非碱性氮化合物甲基衍生物的总离流色谱图;曲线b为通过GC-MS检测的质核比为181的质量色谱图,对应N-甲基咔唑化合物;曲线c为通过GC-MS检测的质核比为195的质量色谱图,对应C1-N-甲基咔唑化合物;曲线d为通过GC-MS检测的质核比为209的质量色谱图,对应C2-N-甲基咔唑化合物;曲线e为通过GC-MS检测的质核比为223的质量色谱图,对应C3-N-甲基咔唑化合物;曲线f为通过GC-MS检测的质核比为237的质量色谱图,对应C4-N-甲基咔唑化合物;曲线g为通过GC-MS检测的质核比为251的质量色谱图,对应C5-N-甲基咔唑化合物。从图1可以看出,色谱图中所有化合物均为9-甲基咔唑类化合物,可见分离得到的非碱性氮化合物甲基衍生物具有很高的纯度。通过色谱质谱分析可以得到非碱性氮化物的分子组成信息。
一种分离与分析石油组分中非碱性氮化合物的方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。













动态评分
0.0