









专利摘要
一种柴油原料的处理方法,包括:将柴油原料油切割成轻柴油馏分和重柴油馏分,所得的轻柴油馏分在第一反应区进行反应,得到富含环烷烃的组分,重柴油馏分在第二反应区反应,所得的加氢重柴油馏分进入催化裂化单元,在催化裂化催化剂的存在下进行催化裂化反应,反应油气经过分离后得到富含芳烃族化合物的汽油产品。本发明可以处理高芳烃含量的柴油原料油,生产BTX、高辛烷值汽油、喷气燃料组分,实现了劣质柴油转化成高附加值的产品的目的。
权利要求
1.一种柴油原料的处理方法,包括:
(1)将柴油原料油切割成轻柴油馏分和重柴油馏分;以轻柴油馏分中总芳烃含量为基准,轻柴油馏分中单环芳烃含量为70%以上,其中烷基苯占单环芳烃含量的60%以上;以重柴油馏分中总芳烃含量为基准,重柴油馏分中多环芳烃含量为80%以上;
(2)步骤(1)所得的轻柴油馏分在第一反应区与加氢精制催化剂I接触,在加氢反应条件下进行反应,第一反应区流出物进行分离,得到富含环烷烃的组分;
(3)步骤(1)所得的重柴油馏分在第二反应区与加氢精制催化剂II接触,在加氢反应条件下进行反应,第二反应区流出物进行分离,得到加氢重柴油馏分,以重柴油馏分为基准,所述加氢重柴油馏分中多环芳烃饱和率达85%以上,生产的单环芳烃选择性达80%以上;
(4)步骤(3)所得的加氢重柴油馏分进入催化裂化单元,在催化裂化催化剂的存在下进行催化裂化反应,反应油气经过分离后得到富含芳烃族化合物的汽油产品。
2.按照权利要求1所述的方法,其特征在于,所述的柴油原料油的沸点范围为150~400℃,总芳烃含量60~90质量%,其中多环芳烃含量40~80质量%。
3.按照权利要求1或2所述的方法,其特征在于,所述的柴油原料油选自催化裂化轻循环油、催化裂化重循环油、环烷基原油的直馏柴油、环烷基原油的焦化柴油、煤直接液化油的柴油馏分、煤焦油的柴油馏分中的一种或几种。
4.按照权利要求1所述的方法,其特征在于,所述的第一反应区的加氢反应条件为:氢分压4~12MPa,反应温度300~400℃,氢油体积比400~1500Nm
5.按照权利要求1所述的方法,其特征在于,所述的第一反应区的加氢反应条件为:氢分压6~12MPa,反应温度320~380℃,氢油体积比400~1000Nm
所述的第二反应区的加氢反应条件为:氢分压6~12MPa,反应温度260~360℃,氢油体积比600~1500Nm
6.按照权利要求1的方法,其特征在于,加氢精制催化剂I中含有载体和负载在所述载体上的钴和钼,以所述加氢精制催化剂I的总重量为基准,以氧化物计,钴的含量为1~10重量%,钼的含量为5~45重量%。
7.按照权利要求6的方法,其特征在于,所述载体选自γ-氧化铝、氧化硅、氧化铝-氧化硅中的一种或多种。
8.按照权利要求7的方法,其特征在于,所述载体为氧化铝-氧化硅。
9.按照权利要求1所述的方法,其特征在于,加氢精制催化剂II中含有载体和负载在所述载体上的加氢金属活性组分;以所述加氢精制催化剂II的总重量为基准,以氧化物计,加氢金属活性组分的含量为15~60重量%。
10.按照权利要求9所述的方法,其特征在于,所述加氢金属活性组分为至少一种选自第VIB族金属元素和至少一种选自第VIII族金属元素,所述第VIB族金属元素为钼和/或钨,所述第VIII族金属元素为钴和/或镍;所述载体选自γ-氧化铝、氧化硅、氧化铝-氧化硅、氧化钛、氧化镁、氧化硅-氧化镁、氧化硅-氧化锆、氧化硅-氧化钍、氧化硅-氧化铍、氧化硅-氧化钛、氧化钛-氧化锆、氧化硅-氧化铝-氧化钍、氧化硅-氧化铝-氧化钛、氧化硅-氧化铝-氧化镁和氧化硅-氧化铝-氧化锆中的一种或多种。
11.按照权利要求1或9所述的方法,其特征在于,所述的加氢精制催化剂II制备方法包括:
(1)采用浸渍法将加氢金属活性组分的水溶性盐和有机络合剂负载到载体上,然后进行干燥、焙烧,得到半成品催化剂,所述焙烧条件使得以半成品催化剂的总量为基准,半成品催化剂中炭含量为0.03-0.5重量%;
(2)以含有有机络合剂的溶液作为浸渍液,对步骤(1)所得半成品催化剂进行浸渍,然后进行干燥且不进行焙烧。
12.按照权利要求11所述的方法,其特征在于,加氢精制催化剂II的制备方法的步骤(1)中所述焙烧在通入气体的条件下进行,且焙烧的温度为350~500℃,焙烧的时间为0.5~8h,气体的通入量为0.2~20升/(克·小时)。
13.按照权利要求12所述的方法,其特征在于,焙烧的温度为360~450℃,焙烧的时间为1~6h,气体的通入量为0.3~10升/(克·小时)。
14.按照权利要求11的方法,其特征在于,加氢精制催化剂II的制备方法的步骤(1)中,有机络合剂与金属活性组分的摩尔比为0.03~2:1。
15.按照权利要求14的方法,其特征在于,加氢精制催化剂II的制备方法的步骤(1)中,有机络合剂与金属活性组分的摩尔比为0.08~1.5:1。
16.按照权利要求11的方法,其特征在于,加氢精制催化剂II的制备方法的步骤(1)所述有机络合剂与步骤(2)所述有机络合剂相同或不同,且所述有机络合剂选自含氧和/或含氮有机物中的一种或多种,所述含氧的有机物选自有机醇、有机酸中的一种或多种,含氮的有机物选自有机胺、有机铵盐中的一种或多种。
说明书
技术领域
本发明涉及采用加氢精制和催化裂化联合处理烃油的方法,更具体地说,是一种柴油原料的处理方法。
背景技术
随着市场对清洁油品需求的快速增长,在我国催化裂化工艺作为生产轻质油品的主要工艺发展迅速,同时催化裂化工艺生产的催化柴油产量逐年增加,催化柴油约占我国商品柴油份额的三分之一。而由于原油重质化,催化柴油质量变差,主要表现为芳烃、硫、氮、烯烃含量高、十六烷值低和安定性差,其中总芳烃含量从50质量%到90质量%之间含量不等,双环和双环以上芳烃含量在40质量%以上,多环芳烃的含量较高是造成催化柴油十六烷值低的主要原因,而环保法规对柴油的质量尤其是十六烷值提出了更为严格的要求,因而导致催化裂化柴油产品难以满足市场质量的要求。此外,随着国家对炼化企业提出降低柴汽比来匹配市场的需求,将劣质柴油加工成高附加值的产品尤为重要。
利用劣质柴油生产高附加值油品的工艺主要有加氢精制和加氢裂化两类。采用常规的加氢精制工艺处理劣质柴油,虽然可以有效地脱除柴油中的硫、氮等杂质,但柴油产品十六烷值提高幅度有限,柴油密度变化也不大,无法满足劣质柴油改质的目的;采用传统的加氢裂化工艺加工劣质柴油,虽然可以生产产品性能优良的低硫柴油调和组分,但需要较为苛刻的反应条件,石脑油馏分收率低且氢耗较大。或者以生产石脑油为目的的加氢裂化工艺,但生产的石脑油馏分辛烷值较低,其研究法辛烷值仅有75 左右。
针对轻循环油(LCO)高芳烃含量的特性,通过控制芳烃转化途径,将LCO中大分子芳烃转化为苯、甲苯、二甲苯(BTX)等小分子芳烃并保持在汽油馏分中,最终生产得到高辛烷值汽油组分或BTX是一条较为理想的LCO高效利用途径。以LCO为原料生产高辛烷值汽油组分或芳烃领域,国外技术主要包括UOP开发的LCO-Unicracking和LCO-X技术、 Criterion催化剂公司开发的一段串联加氢裂化工艺技术和SYN系列柴油加氢改质工艺技术、IFP开发的灵活加氢裂化工艺。国内技术主要包括 RIPP开发的LCO加氢裂化生产高辛烷值汽油(RLG)技术、FRIPP开发的FD2G技术以及RIPP开发的LCO加氢-催化组合技术生产高辛烷值汽油(LTAG)技术。
CN 1422327A公开了一种由LCO多产丙烯的加氢-催化裂化组合方法。该方法是将催化裂化原料经过第一套催化裂化装置,将得到的产物分离得到气体、石脑油、柴油以及重组分。将循环油经过加氢处理后作为原料进入第二套催化裂化装置,提高丙烯产率。该发明要求加氢循环油中十氢化萘含量最大化,且总芳烃含量小于5%(重),在催化裂化过程中容易发生开环裂化反应,生产丙烯。
US6900365公开了一种将LCO通过加氢脱烷基/重整生产BTX或高辛烷值汽油组分的方法。该方法先将LCO加氢处理,得到硫含量、氮含量小于1μg/g的中间产物,然后将该中间产物作为加氢脱烷基/重整原料,经过脱烷基化反应/重整反应得到高辛烷值汽油组分或BTX。该方法中加氢处理单元的主要目的是脱除硫、氮等杂质,防止后面的催化剂中毒。
US6900365公开了一种将LCO通过加氢脱烷基/重整生产BTX或高辛烷值汽油组分的方法。该方法先将LCO加氢处理,得到硫含量、氮含量小于1μg/g的中间产物,然后将该中间产物作为加氢脱烷基/重整原料,经过脱烷基化反应/重整反应得到高辛烷值汽油组分或BTX。该方法中加氢处理单元的主要目的是脱除硫、氮等杂质,防止后面的催化剂中毒。
CN 101942330 A公开了一种由催化裂化柴油加氢的方法,将催化裂化柴油分离成沸点小于230℃的组分和沸点为230℃以上的组分,然后使沸点为230℃以上的组分中的多环芳烃加氢精制为单环芳烃,将得到的富含单环芳烃的柴油进行催化裂化。本发明的方法可以提高加氢选择性,制得芳烃含量和辛烷值高的FCC汽油。由于双环芳烃,如萘的沸点为218℃,按照上述要求仅将230℃以上的组分进行选择性加氢饱和,会引起单环芳烃收率的降低。
发明内容
本发明所要解决的技术问题是高芳烃柴油原料在常规加氢精制过程中的单环芳烃选择性差的问题。
无论用高芳烃柴油原料加氢裂化生产高辛烷值汽油组分,还是用高芳烃柴油原料加氢—催化裂化组合生产高辛烷值汽油组分或芳烃,都是先将高芳烃柴油原料加氢精制,然后作为加氢裂化或催化裂化的进料,其加氢精制的目的就是将高芳烃柴油原料中的多环芳烃选择性加氢为单环芳烃。
高芳烃柴油原料中芳烃主要包括单环芳烃、双环芳烃和三环以上芳烃。本发明的发明人深入研究发现,芳烃加氢是可逆反应,受动力学和热力学双重控制,在较低反应温度区间,芳烃加氢饱和反应主要受动力学控制,在较高反应温度区间,芳烃加氢饱和反应主要受热力学控制。反应工艺参数对芳烃加氢产生复杂的影响。一方面,在动力学控制区提高温度有利于提高反应速率;另一方面,温度提高到一定程度后,将导致热力学平衡的限制。现有技术中,在加氢精制过程,不论是未将单环芳烃作为主要的目标产物,还是将单环芳烃作为主要目标产物,都出现了芳烃过度饱和的问题,导致最终单环芳烃含量下降,即单环芳烃选择性差的问题,同时还存在氢耗增加等问题。
在本发明中,多环芳烃是指双环芳烃和三环以上芳烃(包括三环芳烃) 的总称。
本发明的发明人进一步深入研究发现,多环芳烃的第一个环加氢饱和反应通常比单环芳烃加氢饱和反应速率常数大,也就是说多环芳烃第一个环的加氢较单环芳烃的加氢饱和容易。为了提高多环芳烃的饱和率,需要适当提高反应苛刻度,包括提高反应温度,但是提高反应温度,又会使单环芳烃加氢饱和,如果进一步提高反应温度,则芳烃加氢饱和反应会进入热力学控制区域,多环芳烃的饱和率反而会下降。本发明的发明人通过研究,获得了单环芳烃的选择性与多环芳烃的饱和率关系图,参见附图2。在较低的反应温度下,随着反应温度的升高,多环芳烃饱和率增加,且饱和的多环芳烃全部转化为单环芳烃,此时单环芳烃的选择性为100%,当反应温度上升到一定值,继续提高反应温度,多环芳烃饱和率继续上升,但此时开始发生单环芳烃的加氢饱和转化为环烷烃的反应,单环芳烃的选择性开始下降,当反应温度达到动力学控制临界值时,此时多环芳烃饱和率达到最大值,单环芳烃选择性达到最低值,继续提高反应温度,芳烃加氢饱和反应进入热力学控制区域,在热力学控制区域提高反应温度,多环芳烃饱和率下降,单环芳烃选择性上升。
基于上述研究,本发明所要解决的技术问题是高芳烃柴油原料在常规加氢精制过程中的单环芳烃选择性差的问题。本发明提供一种以高芳烃含量柴油为原料生产BTX、高辛烷值汽油、喷气燃料组分的方法。
本发明提供了一种柴油原料的处理方法,包括:(1)将柴油原料油切割成轻柴油馏分和重柴油馏分;
(2)步骤(1)所得的轻柴油馏分在第一反应区与加氢精制催化剂I 接触,在加氢反应条件下进行反应,第一反应区流出物进行分离,得到富含环烷烃的组分;
(3)步骤(1)所得的重柴油馏分在第二反应区与加氢精制催化剂II 接触,在加氢反应条件下进行反应,第二反应区流出物进行分离,得到加氢重柴油馏分,以重柴油馏分为基准,所述加氢重柴油馏分中多环芳烃饱和率达85%以上,生产的单环芳烃选择性达80%以上;
(4)步骤(3)所得的加氢重柴油馏分进入催化裂化单元,在催化裂化催化剂的存在下进行催化裂化反应,反应油气经过分离后得到富含芳烃族化合物的汽油产品。
所述的柴油原料油的沸点范围为150~400℃,总芳烃含量60~90质量%,其中多环芳烃含量40~80质量%。优选,柴油原料油的总芳烃含量高于70质量%,其中双环以上芳烃的含量高于45质量%,氮含量为100 μg.g
本发明为了提高多环芳烃的饱和率,并抑制单环芳烃的加氢饱和,根据单环芳烃和多环芳烃的分布特点,本发明将上述柴油原料油按照烃类组成进行轻、重切割,然后通过两个反应区,对不同馏分分别进行不同深度的加氢精制反应。以轻柴油馏分中总芳烃含量为基准,轻柴油馏分中单环芳烃含量为70%以上,其中烷基苯占单环芳烃含量的60%以上;以重柴油馏分中总芳烃含量为基准,重柴油馏分中多环芳烃含量为80%以上。
所得轻柴油馏分在第一反应区与加氢精制催化剂I接触,在加氢反应条件下进行反应,以轻柴油馏分为基准,所得轻柴油馏分中芳烃饱和率达到90%以上。第一反应区流出物进行分离,得到富含环烷烃的组分,该组分是优质的喷气燃料组分。所得的重柴油馏分在第二反应区与加氢精制催化剂II接触,在加氢反应条件下进行反应,第二反应区流出物进行分离,得到加氢重柴油馏分,以重柴油馏分为基准,所述加氢重柴油馏分中多环芳烃饱和率达85%以上,生产的单环芳烃选择性达80%以上。在本发明中:
多环芳烃饱和率及单环芳烃选择性定义如下:
多环芳烃饱和率=(Adf-Adp)/Adf*100%
单环芳烃选择性=(Amp-Amf)/(Adf-Adp)*100%
式中:Adf—原料中多环芳烃含量,质量%
Adp—产品中多环芳烃含量,质量%
Amp—产品中单环芳烃含量,质量%
Amf—原料中单环芳烃含量,质量%
本发明所述的“多环芳烃含量”指的是以质谱法(分析方法SH/T-0606) 得到的质谱组成数据中双环以上芳烃其中包括双环芳烃本身的质量分数之和。
本发明所述的“单环芳烃含量”指的是以质谱法(分析方法SH/T-0606) 得到的质谱组成数据中单环芳烃的质量分数。
优选,本发明中所述的第一反应区的加氢反应条件为:氢分压 4~12MPa,反应温度300~400℃,氢油体积比400~1500Nm
优选,本发明中所述的第二反应区的加氢反应条件为:氢分压 5~10MPa,反应温度240~380℃,氢油体积比400~1500Nm
在本发明其中一种优选实施方式中,加氢精制催化剂I中含有载体和负载在所述载体上的钴和钼,以所述加氢精制催化剂I的总重量为基准,以氧化物计,钴的含量为1~10重量%,钼的含量为5~45重量%。
优选,所述载体选自γ-氧化铝、氧化硅、氧化铝-氧化硅中的一种或多种,进一步优选,所述载体为氧化铝-氧化硅。
在本发明其中一种优选实施方式中,加氢精制催化剂II中含有载体和负载在所述载体上的加氢金属活性组分;以所述加氢精制催化剂II的总重量为基准,以氧化物计,加氢金属活性组分的含量为15~60重量%。优选为20~50重量%。
优选所述加氢金属活性组分为至少一种选自第VIB族金属元素和至少一种选自第VIII族金属元素,所述第VIB族金属元素优选为钼和/或钨,所述第VIII族金属元素优选为钴和/或镍;所述载体选自γ-氧化铝、氧化硅、氧化铝-氧化硅、氧化钛、氧化镁、氧化硅-氧化镁、氧化硅-氧化锆、氧化硅-氧化钍、氧化硅-氧化铍、氧化硅-氧化钛、氧化硅-氧化锆、氧化钛-氧化锆、氧化硅-氧化铝-氧化钍、氧化硅-氧化铝-氧化钛、氧化硅-氧化铝-氧化镁和氧化硅-氧化铝-氧化锆中的一种或多种。
在本发明其中一种优选实施方式中,所述的加氢精制催化剂II由下述制备方法制备获得,所述制备方法包括:
(1)采用浸渍法将加氢金属活性组分的水溶性盐和有机络合剂负载到载体上,然后进行干燥、焙烧,得到半成品催化剂,所述焙烧条件使得以半成品催化剂的总量为基准,半成品催化剂中炭含量为0.03-0.5重量%;
(2)以含有有机络合剂的溶液作为浸渍液,对步骤(1)所得半成品催化剂进行浸渍,然后进行干燥且不进行焙烧。
在本发明中,优选的加氢精制催化剂II为两步浸渍法制备催化剂,第一步浸渍和第二部浸渍分别用于引入加氢金属活性组分和有机络合剂,在第一步浸渍过程中加入有机络合剂并使之通过焙烧转化为炭,不仅能够提高催化剂的活性,而且能够有效地长时间保持催化剂的高活性,从而大大提高催化剂的使用寿命。推测其原因可能是因为第一步浸渍过程中加入的有机络合剂,有机络合剂的存在阻碍了焙烧过程中活性金属的聚集,使其分散的更加均匀;同时,第一步浸渍后焙烧能够使金属化合物转化为金属氧化物,使有机络合剂转化为炭,从而使活性金属与载体之间的结合更加牢固,提高了催化剂的活性和稳定性。而在第二步浸渍过程中加入的有机络合剂覆盖在催化剂表面,能够有效防止活性金属在硫化过程中的聚集,提高金属分散度,更有利于形成具有更高活性的Ⅱ类活性相以及形成更多的活性中心,从而进一步挺高了催化剂的活性。因此,该技术可有效解决常规浸渍法与现有络合浸渍法的技术缺陷。
优选的,本发明提供的加氢精制催化剂II的制备方主要包括以下步骤:
(1)采用浸渍法将加氢金属活性组分的水溶性盐和有机络合剂负载到载体上,然后进行干燥、焙烧,得到半成品催化剂,所述焙烧条件使得以半成品催化剂的总量为基准,优选地,半成品催化剂中炭含量为0.04~0.4 重量%。
(2)以含有有机络合剂的溶液作为浸渍液,对步骤(1)所得半成品催化剂进行浸渍,然后进行干燥且不进行焙烧。
在本发明中,可以通过控制焙烧条件中的焙烧温度和可燃性气体的通入量来获得上述炭含量,所述可燃性气体可以为各种氧气含量不低于20 体积%的气体,如空气、氧气以及它们的混合气体中的一种或多种。
所述可燃性气体的通入量不低于0.2升/克·小时。所述可燃性气体的通入,一方面满足燃烧的条件,使得活性金属组分的盐转化为氧化物,使有机络合剂转化为炭;另一方面也能将燃烧形成的二氧化碳和水以及其他成分排放出去,以避免沉积在催化剂上造成对活性相的空位阻碍。
优选情况下,可燃性气体的通入量为0.2~20升/(克·小时),优选为 0.3~10升/(克·小时)。此处的“克”表示载体的重量。
根据本发明,优选地,步骤(1)所述焙烧的温度为350~500℃,优选为360~450℃,焙烧的时间为0.5~8h,优选为1~6h。控制焙烧温度在上述范围内即可保证有机络合剂能以上述含量范围将炭形成于载体上,得到半成品催化剂。
根据本发明,优选地,步骤(1)所述有机络合剂与以金属活性组分的摩尔比为0.03~2:1,优选为0.08~1.5:1。
根据本发明,优选地,步骤(1)和步骤(2)有机络合剂的摩尔比为 1:0.25~4,优选为1:0.5~2。
在本发明中,步骤(1)和步骤(2)中所述有机络合剂可以相同,也可以不同,优选地,所述有机络合剂选自含氧和/或含氮有机物中的一种或多种。
所述含氧的有机物优选选自有机醇、有机酸中的一种或多种。
所述有机醇优选为二元以上的多元醇,更进一步优选为碳原子数2-6 的多元醇或其低聚体或多聚体,如乙二醇、丙三醇、聚乙二醇、二乙二醇、丁二醇中的一种或多种。所述聚乙二醇的分子量优选为200~1500。
所述有机酸优选为C2-C7的含一个或多个COOH基团的化合物,具体可以为乙酸、马来酸、草酸、氨基三乙酸、1,2-环己烷二胺四乙酸、柠檬酸、酒石酸、苹果酸中的一种或多种。
所述含氮的有机物优选选自有机胺、有机铵盐中的一种或多种。
所述有机胺优选为C2-C7的含一个或多个NH基团的化合物,可以是伯胺、仲胺或叔胺,特别优选为乙二胺。
所述有机铵盐优选为EDTA。
具体地,本发明特别优选所述有机络合剂为乙二醇、丙三醇、聚乙二醇(分子量优选为200-1500)、二乙二醇、丁二醇、乙酸、马来酸、草酸、氨基三乙酸、1,2-环己烷二胺四乙酸、柠檬酸、酒石酸、苹果酸、乙二胺和EDTA中的一种或多种。
优选地,步骤(1)中所述有机络合剂选自有机酸中的一种或多种,更优选地,步骤(1)所述有机络合剂选自C2-C7的有机酸中的一种或多种。使用有机酸作为步骤(1)的有机络合剂,可以获得具有更高活性的加氢催化剂。
本发明对所述干燥条件没有特别的限定,可以是本领域常用的各种干燥条件,步骤(1)和步骤(2)中所述干燥条件可以相同,也可以不同。
优选地,步骤(1)所述干燥温度为100~250℃,时间为1~12h。
优选地,步骤(2)所述干燥温度为100~200℃,时间为1~12h。
根据本发明,优选地,以金属元素计,加氢金属活性组分的水溶性盐的浓度为0.2~8mol/L,优选为0.2~5mol/L,更进一步优选为0.2~2mol/L。此处的浓度为各种加氢金属活性组分的水溶性盐各自的浓度,而非总浓度。
根据本发明,优选地,含第VIB族金属元素的化合物可以选自钼酸铵、仲钼酸铵、偏钨酸铵、氧化钼和氧化钨中的一种或多种。
优选地,含第VIII族金属元素的化合物选自第VIII族金属的草酸盐、第VIII族金属的硝酸盐、第VIII族金属的硫酸盐、第VIII族金属的醋酸盐、第VIII族金属的氯化物、第VIII族金属的碳酸盐、第VIII族金属的碱式碳酸盐、第VIII族金属的氢氧化物、第VIII族金属的磷酸盐、第VIII 族金属的钼酸盐、第VIII族金属的钨酸盐和第VIII族金属的水溶性氧化物中的一种或多种。
进一步优选,含第VIII族金属元素的化合物可以选自但不限于硝酸镍、硫酸镍、醋酸镍、碱式碳酸镍、硝酸钴、硫酸钴、醋酸钴、碱式碳酸钴、氯化钴和氯化镍中的一种或多种。
根据本发明,对所述加氢金属活性组分的负载方式没有特别的限制。
根据本发明,优选地,所述加氢金属活性组分的负载是通过浸渍法将加氢金属活性组分负载到载体上。
步骤(3)所得的加氢重柴油馏分进入催化裂化单元,在催化裂化催化剂的存在下进行催化裂化反应,反应油气经过分离后得到干气、液化气、汽油、柴油和油浆,所得汽油产品富含芳烃族化合物,为高辛烷值汽油产品。催化裂化单元的反应条件、催化裂化催化剂以及其他再生和分离过程均是本领域常规的手段或方法。
本发明的优点:
(1)本发明根据高芳烃含量柴油原料的烃类组成分布进行切割,根据组成分布进行不同深度的加氢精制,提高了氢气资源的有效利用率,提高了过程的经济性
(2)采用本发明提供的方法,可以处理高芳烃含量的柴油原料油,生产BTX、高辛烷值汽油、喷气燃料组分。通过本方法可以得到符合 GB6537-2006 3号喷气燃料标准的产品,同时为催化裂化生产高辛烷值汽油或者BTX组分提供了优质原料,降低了加工催化柴油的总氢耗,。与现有技术相比,在保持高的多环芳烃饱和率的同时具有高的单环芳烃选择性,多环芳烃饱和率达85%以上时,单环芳烃选择性达80%以上。实现了劣质柴油转化成高附加值的产品的目的。
附图说明
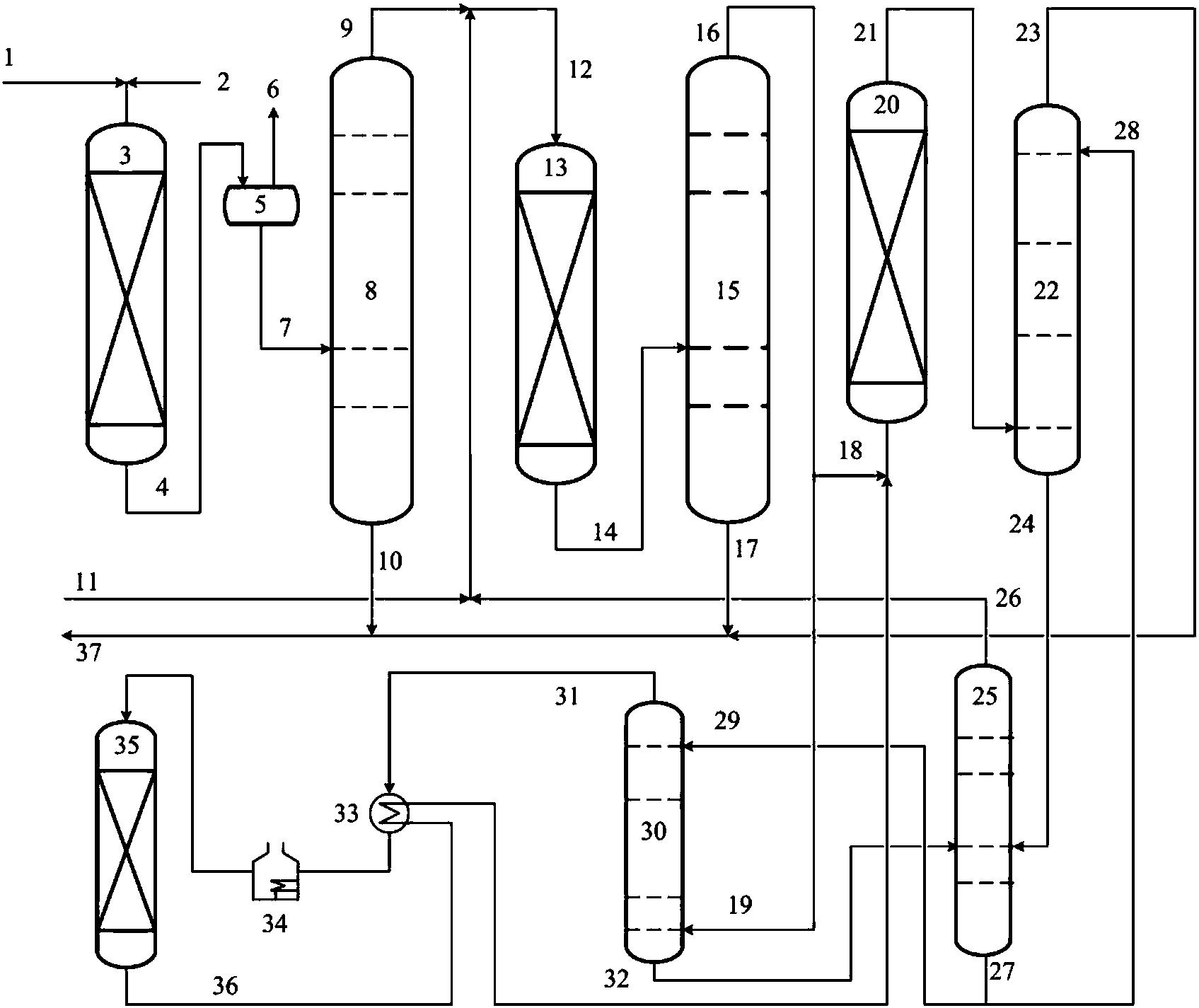
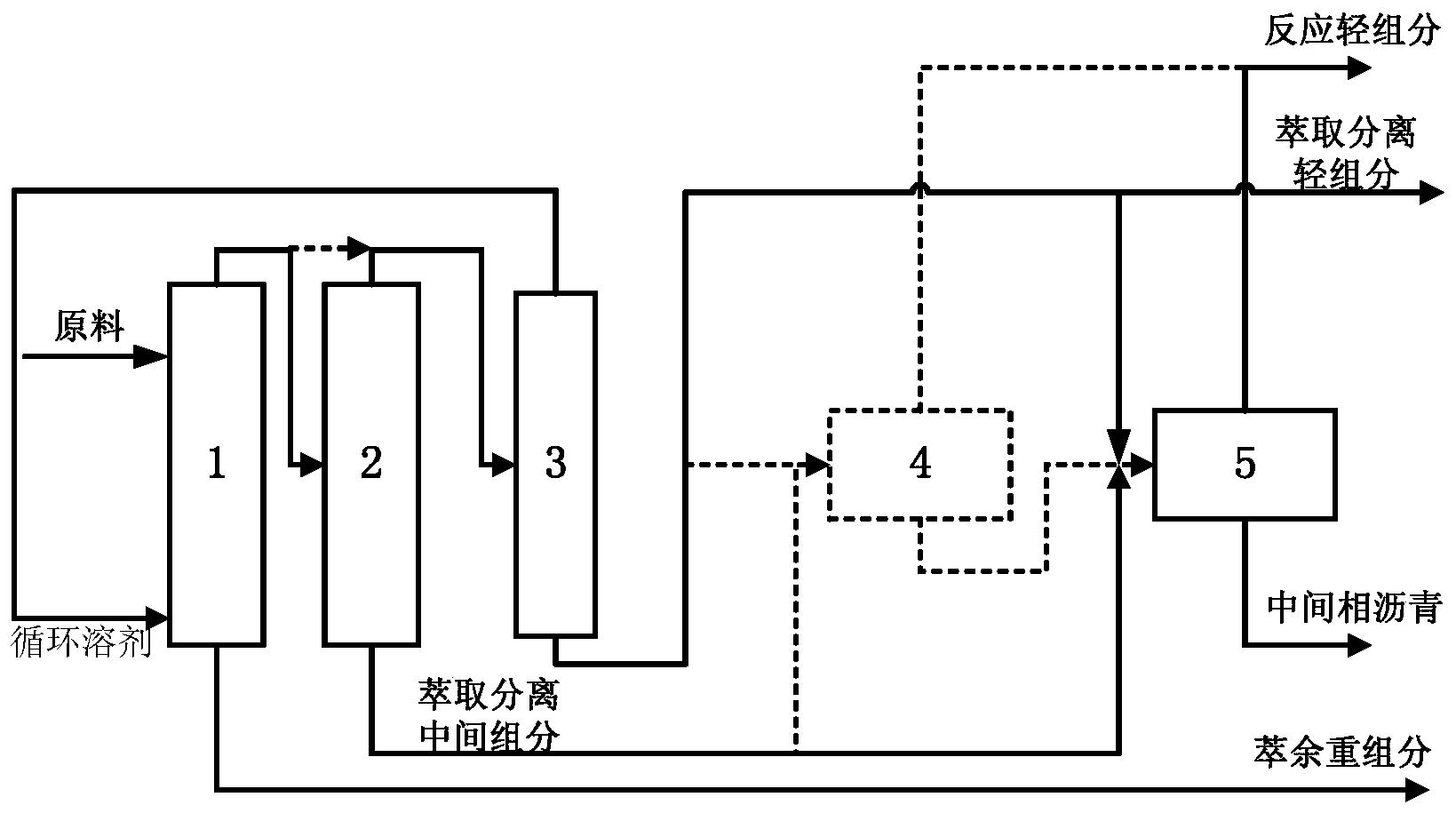
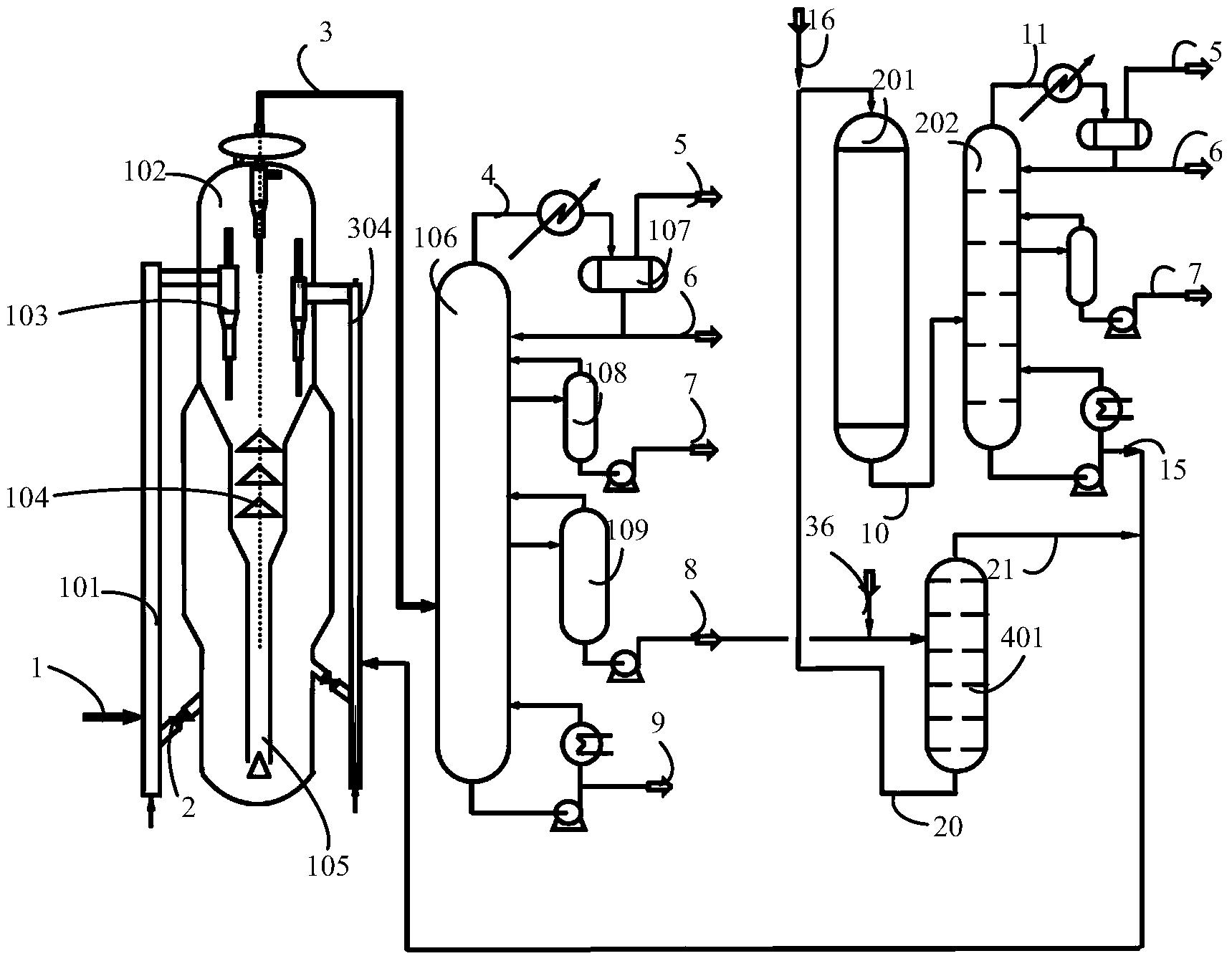
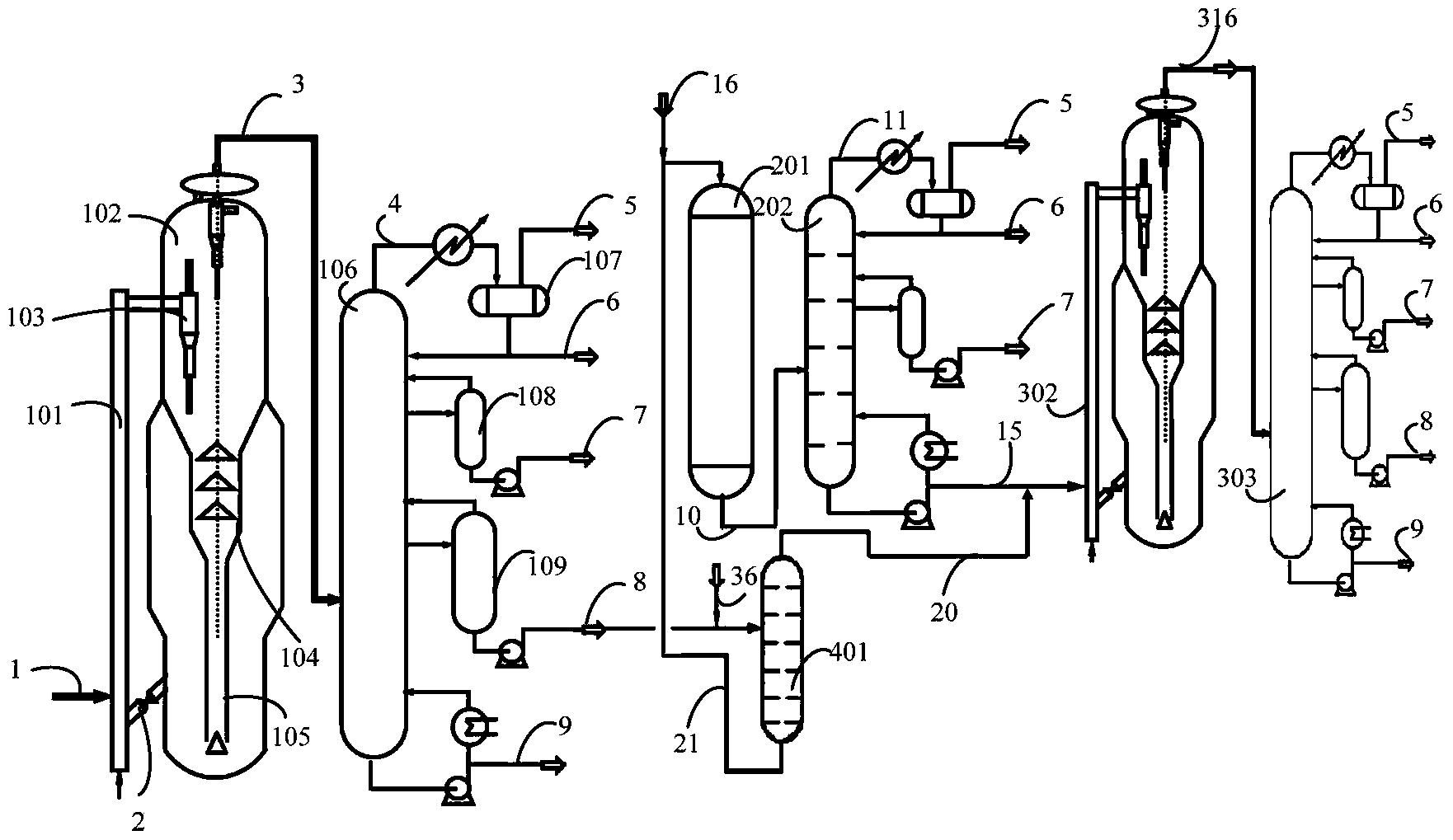
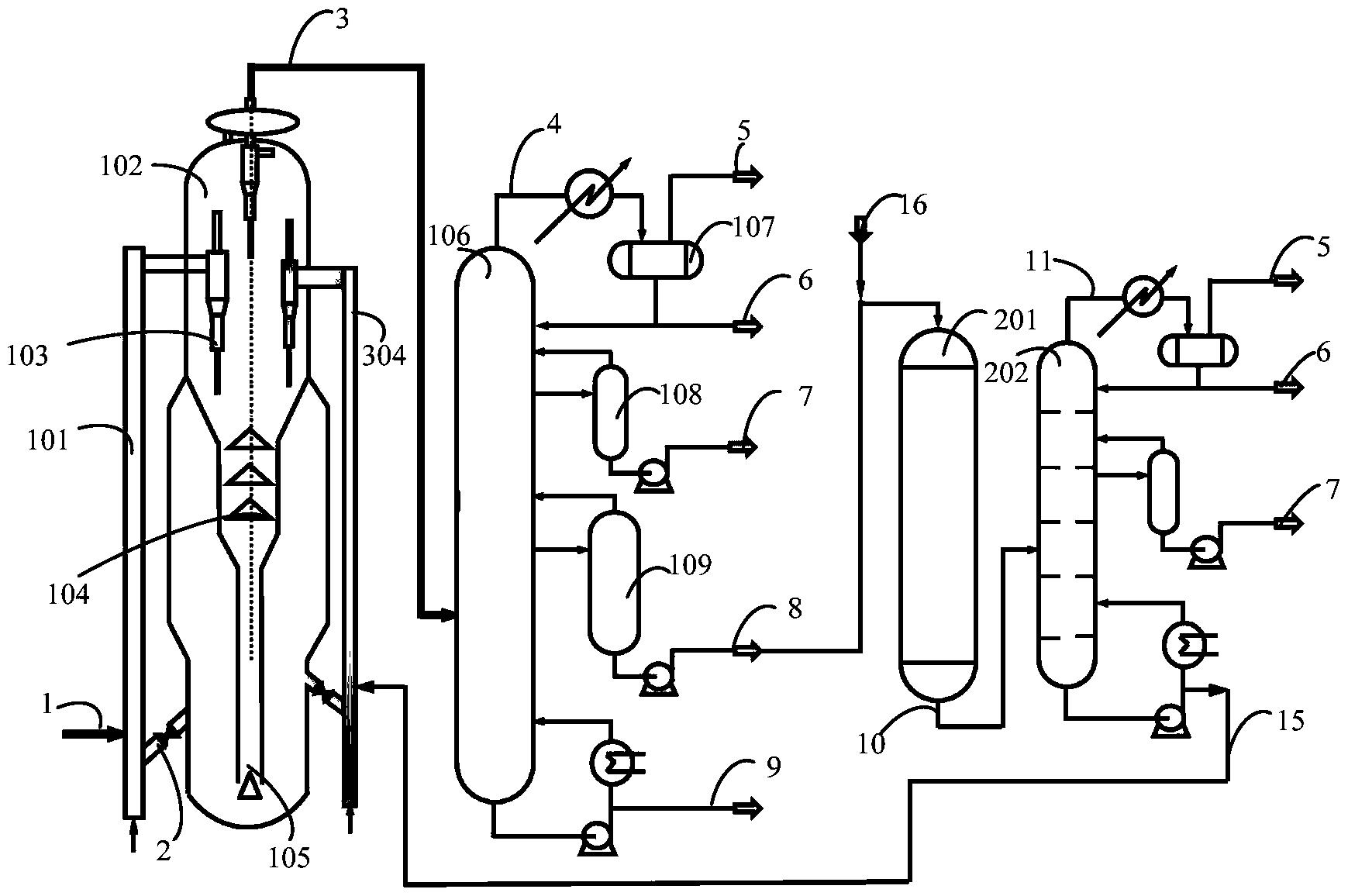
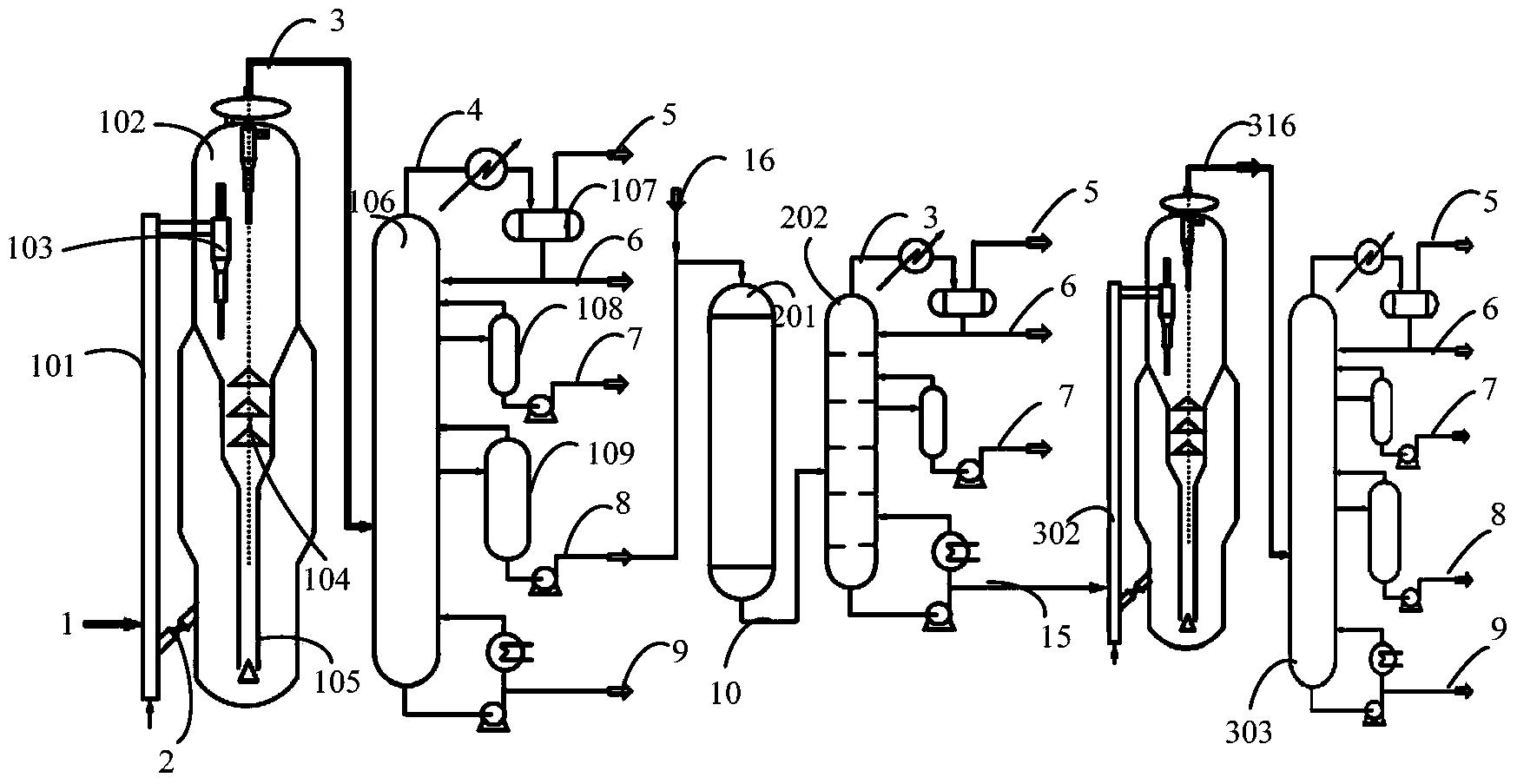
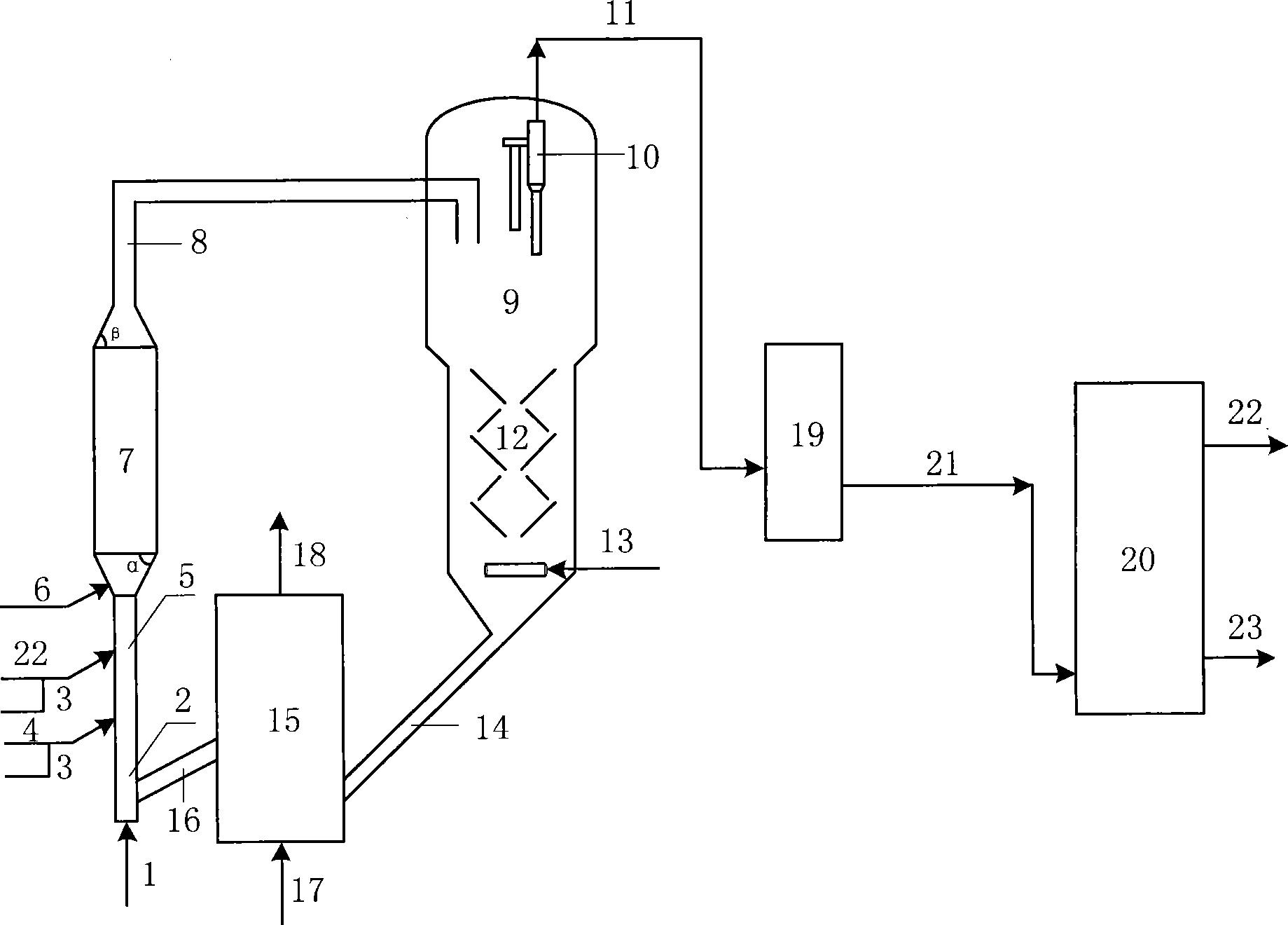
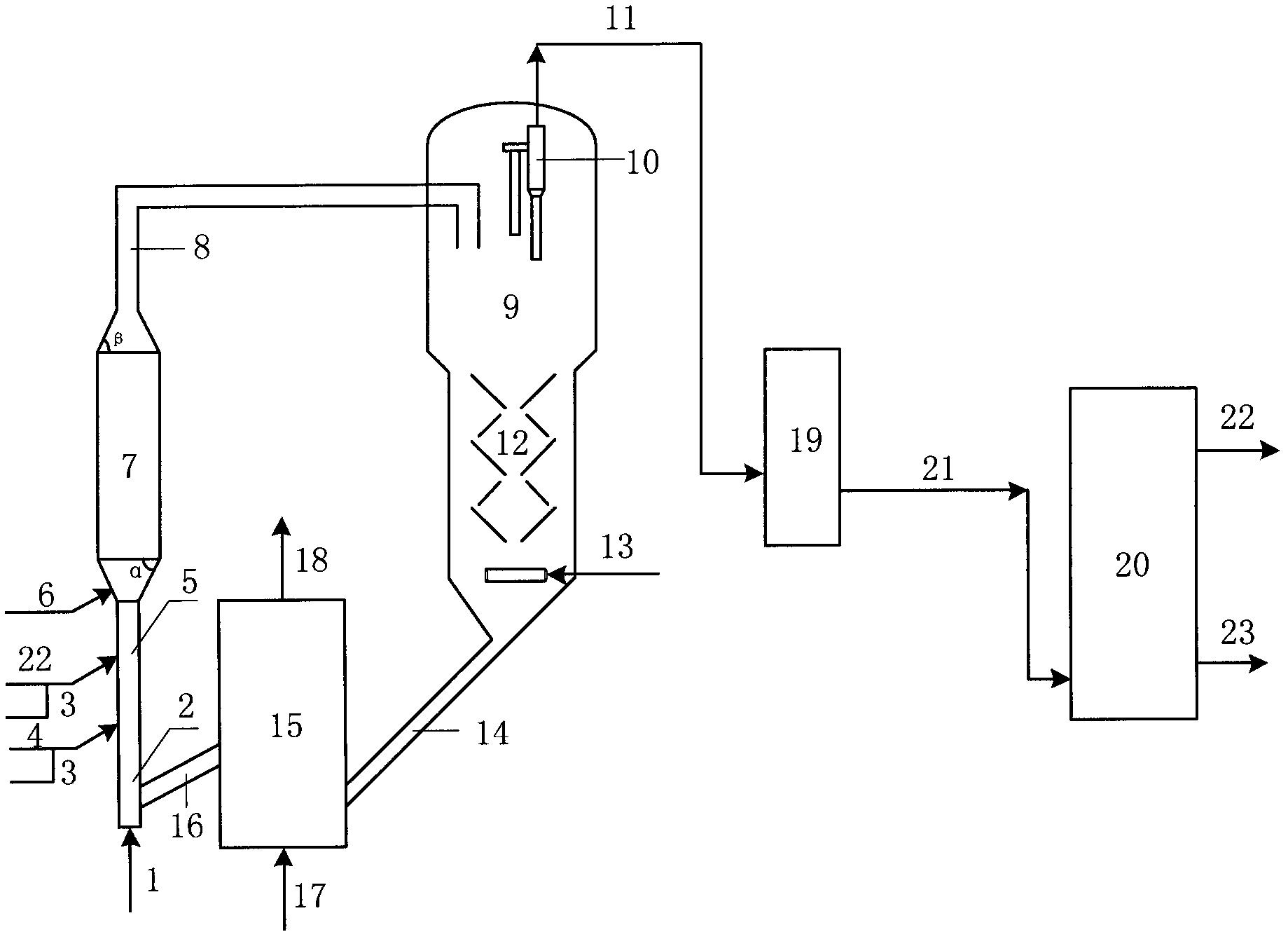
图1是本发明提供柴油处理方法其中一种实施方式的流程示意图。
图2是单环芳烃的选择性与多环芳烃的饱和率关系图。
具体实施方式
下面结合附图对本发明所提供的方法进行进一步的说明,但并不因此而限制本发明。
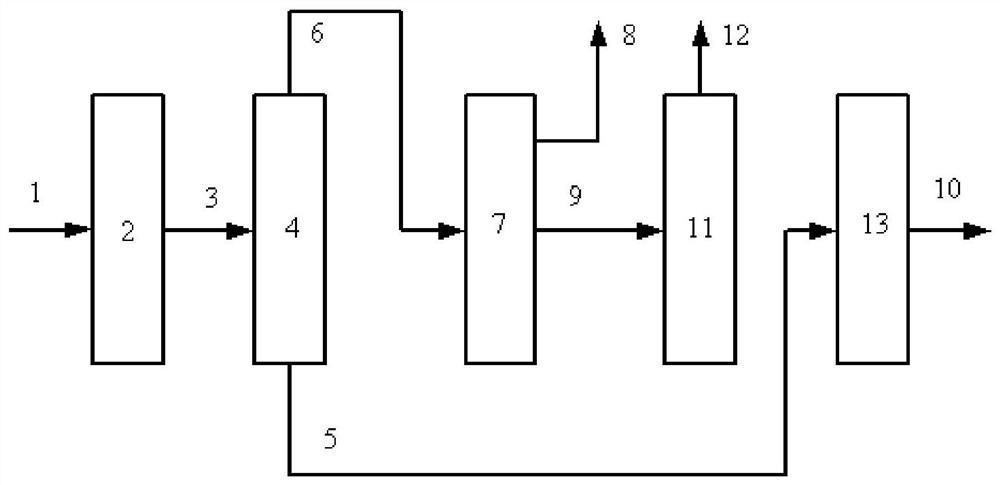
图1是本发明提供方法其中一种实施方式的流程示意图。如图1所示,来自管线1的柴油原料油经过分馏塔2分为轻柴油馏分和重柴油馏分,轻柴油馏分经过管线4与来自循环氢压缩机10经过管线11输送的氢气混合,同时根据系统压力情况可以通过管线12进行补充新氢,混合后的油气混合物经管线5进入加氢反应器6,与加氢精制催化剂I接触进行反应。反应后的物流经过管线7进入气液分离器8。分离的气相物流通过管线9进入循环氢压缩机10;分离的液相物流通过管线13进入分馏塔14,分离得到喷气燃料组分经管线15抽出,少量的石脑油馏分经管线16抽出。重柴油馏分经过管线3与来自循环氢压缩机20经过管线21输送的氢气混合,同时根据系统压力情况可以通过管线22进行补充新氢,混合后的油气混合物进入加氢反应器17,与加氢精制催化剂II接触进行反应。加氢反应器17可以根据实际情况设置多个床层,并且在床层间可以设置补充冷氢。经过加氢反应器17的反应后物流经过管线18进入气液分离器24。分离的气相物流通过管线19进入循环氢压缩机20;分离的液相物流通过管线25 进入分馏塔26,分离得到部分石脑油馏分经管线27输送出去,得到的加氢重柴油馏分经管线28进入催化裂化单元29。在催化裂化催化剂的作用发生催化裂化反应。从催化裂化装置分离得到高辛烷值汽油产品经过管线 31输出。催化裂化柴油由管线30输送至分馏塔2之前。
下面的实施例将对本发明提供的方法予以进一步的说明,但并不因此而限制本发明。
以下实施例和对比例中,催化剂中各元素的含量进行分析,采用商购自日本理学电机工业株式会社的3271E型X射线荧光光谱仪测定。
以下实施例和对比例中,喷气燃料馏分收率定义为全馏分产品通过分馏塔分馏出的喷气燃料馏分与原料油的重量百分比。
实施例中所使用的加氢处理催化剂A、B的制备方法如下:
催化剂A制备方法:分别称取27克三氧化钼、8克碱式碳酸钴、4克磷酸、8克柠檬酸放入140g去离子水中,搅拌得到澄清浸渍溶液S。采用饱和浸渍法用上述溶液浸渍将2000克氢氧化铝粉和1039克硅溶胶混合均匀。将得到的混合物用挤条机挤成外接圆直径为1.4毫米的蝶形条,并将挤出的湿条在120℃干燥4小时,接着在600℃焙烧3小时,制得载体Z,载体Z中氧化硅含量为18.0重量%,氧化铝含量为82.0重量%。Z1的吸水率为0.85。将S溶液定容至85mL,取100克载体Z饱和浸渍2小时,然后依次在120℃干燥2小时,250℃干燥3小时,得到催化剂A,以A 的总量为基准,以氧化物计,CoO含量为4.3重%,MoO3含量为25.1重%。
催化剂B制备方法:分别称取30克硝酸镍、45克偏钨酸铵和15克草酸放入140克去离子水中,搅拌溶解得到澄清溶液,采用饱和浸渍法用上述溶液浸渍200克氧化硅载体,浸渍时间为2小时,然后,在120℃干燥 2小时,接着将其在通入空气流的状态下进行焙烧,焙烧温度为450℃,时间为4小时,气剂比为0.3升/(克·小时),得到半成品催化剂,半成品催化剂的炭含量为0.1重%;将10克二甘醇放入150克去离子水中,搅拌得到澄清溶液,采用饱和浸渍法用上述溶液浸渍半成品催化剂,浸渍时间为2小时,然后,在120℃干燥6个小时,得到催化剂C。以C的总量为基准,以氧化物计,NiO含量为3.3重%,WO3含量为15.1重%。
为充分发挥催化剂的加氢脱硫性能,上述催化剂在接触正式原料前均需要进行预硫化处理。以下所列对比例和实施例中,各催化剂的预硫化方法相同。
实施例、对比例中加氢单元的精制催化剂为中国石化催化剂分公司生产的RS-2000,RN-32L催化剂,催化裂化单元采用的是中国石化催化剂分公司生产的HAC催化剂,上述三种催化剂均已经商业化生产并使用。
实施例、对比例中所使用的原料性质列于表1。
实施例1
按照本发明提供的方法,采用表1中的催化柴油,将其切割成轻柴油馏分A1、重柴油馏分B1,以轻柴油馏分A1中总芳烃含量为基准,切割出的轻柴油馏分A1中单环芳烃含量占总芳烃含量的83.3%,烷基苯占单环芳烃65%;以重柴油馏分B1中总芳烃含量为基准,重柴油馏分B1中双环芳烃占总芳烃含量的87.4%。轻柴油馏分A1在第一反应区与催化剂 A接触进行反应,重柴油馏分B1在第二反应区与催化剂B接触进行反应。其加氢反应条件如表 2所示。加氢重柴油馏分进入催化裂化提升管反应器,在催化裂化催化剂HAC存在下进行催化裂化反应,催化裂化装置的反应条件:反应温度为560℃,剂油质量比为20,油气停留时间为7s,压力为 0.25MPa,水蒸气/原料为0.04,老化剂MAT为66。得到的反应油气经过分离得到包括富含芳烃的汽油、轻循环油等产品。所得到产品具体性质见表4。
第一反应区反应流出物经分离后得到的喷气燃料组分,其烟点为 25.6mm,冰点为-54℃,其他性质符合GB6537-2006 3号喷气燃料标准。第二反应区反应流出物经分离后得到加氢重柴油馏分,与重柴油馏分相比,多环芳烃饱和率为87.2%,单环芳烃选择性为85.5%。将加氢重柴油馏分进入催化裂化单元与不进入催化裂化单元的情况进行对比,加氢重柴油馏分进入催化裂化单元后,催化裂化单元产物中汽油收率提高了13.5%,其辛烷值增加了0.4个单位。
对比例1
采用表1中的催化柴油,将其按照270℃的切割点,切割成轻柴油馏分A2、重柴油馏分B2,以轻柴油馏分A2中总芳烃含量为基准,切割出的轻柴油馏分A2中单环芳烃含量占总芳烃含量的68.1%,其中烷基苯占单环芳烃59%。以重柴油馏分B2中总芳烃含量为基准,重柴油馏分B2 中双环芳烃含量占总芳烃含量的86.6%。轻柴油馏分A2在第一反应区与催化剂A接触进行反应,重柴油馏分B2在第二反应区与催化剂B接触进行反应。其加氢反应条件如表 2 所示。加氢重柴油馏分进入催化裂化提升管反应器,在催化裂化催化剂HAC存在下进行催化裂化反应,催化裂化装置的反应条件:反应温度为560℃,剂油质量比为20,油气停留时间为 7s,压力为0.25MPa,水蒸气/原料为0.04,老化剂MAT为66。得到的反应油气经过分离得到包括富含芳烃的汽油、轻循环油等产品。所得到产品具体性质见表4。
第一反应区反应流出物经分离后得到的喷气燃料组分,其烟点为 23.1mm,冰点为-48℃,其他性质符合GB6537-2006 3号喷气燃料标准。第二反应区反应流出物经分离后得到加氢重柴油馏分,与重柴油馏分相比,多环芳烃饱和率为84.5%,单环芳烃选择性为81.5%。将加氢重柴油馏分进入催化裂化单元与不进入催化裂化单元的情况进行对比,加氢重柴油馏分进入催化裂化单元后,催化裂化单元产物中汽油收率提高了10.1%,其辛烷值增加了0.2个单位。
对比例2
采用表1中的催化柴油,采用与实施例1相同的切割点,将其切割成轻柴油馏分A1、重柴油馏分B1。轻柴油馏分A1在第一反应区与催化剂 RS-2000接触进行反应,重柴油馏分B1在第二反应区与催化剂RN-32L接触进行反应。其加氢反应条件如表 2 所示。加氢重柴油馏分进入催化裂化提升管反应器,在催化裂化催化剂HAC存在下进行催化裂化反应,得到的反应油气经过分离得到包括富含芳烃的汽油、轻循环油等产品。所得到产品具体性质见表4。
第一反应区反应流出物经分离后得到的喷气燃料组分,其烟点为 22mm,冰点为-47℃,其他性质符合GB6537-2006 3号喷气燃料标准。第二反应区反应流出物经分离后得到加氢重柴油馏分,与重柴油馏分相比,多环芳烃饱和率为86%,单环芳烃选择性为60.5%。将加氢重柴油馏分进入催化裂化单元与不进入催化裂化单元的情况进行对比,加氢重柴油馏分进入催化裂化单元后,催化裂化单元产物中汽油收率提高了8.7%,其辛烷值增加了0.2个单位。
对比例3
采用表1中的催化柴油,将其按照230℃的切割点,切割成轻柴油馏分A3、重柴油馏分B3,以轻柴油馏分A3中总芳烃含量为基准,切割出的轻柴油馏分A3中单环芳烃含量占总芳烃含量的94.1%,其中烷基苯占单环芳烃52%。以重柴油馏分B3中总芳烃含量为基准,重柴油馏分B3 中双环芳烃含量占总芳烃含量的68.2%。轻柴油馏分A3在第一反应区与催化剂A接触进行反应,重柴油馏分B3在第二反应区与催化剂B接触进行反应。其加氢反应条件如表 2 所示。加氢重柴油馏分进入催化裂化提升管反应器,在催化裂化催化剂HAC存在下进行催化裂化反应,催化裂化装置的反应条件:反应温度为560℃,剂油质量比为20,油气停留时间为 7s,压力为0.25MPa,水蒸气/原料为0.04,老化剂MAT为66。得到的反应油气经过分离得到包括富含芳烃的汽油、轻循环油等产品。所得到产品具体性质见表4。
第一反应区反应流出物经分离后得到的喷气燃料组分,其烟点为27.1mm,冰点为-57℃,其他性质符合GB6537-2006 3号喷气燃料标准。第二反应区反应流出物经分离后得到加氢重柴油馏分,与重柴油馏分相比,多环芳烃饱和率为86.5%,单环芳烃选择性为65%。将加氢重柴油馏分进入催化裂化单元与不进入催化裂化单元的情况进行对比,加氢重柴油馏分进入催化裂化单元后,催化裂化单元产物中汽油收率提高了9.5%,其辛烷值增加了0.1个单位。
对比例4
采用表1中的催化柴油不经过切割,直接进行加氢精制,采用的是 RS-2000催化剂,加氢精制操作条件如表3所示。其反应流出物进行分离,液体产物经过分馏切割出喷气燃料组分,具体产品性质见表5。所得喷气燃料组分中烟点为23.5mm,冰点为-48℃,其他性质符合GB6537-2006 3 号喷气燃料标准。以原料为基准,液体产物中多环芳烃饱和率为98.0%,单环芳烃选择性为-36.8%,表明为了得到合格的喷气燃料产品,原料进行了过度加氢,大量的单环芳烃进行了加氢饱和,导致氢耗高达为4.2%。
表1原料油性质
表2操作参数
表3
表4实施例及对比例产品性质
表5对比例产品性质
一种柴油原料的处理方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。







动态评分
0.0