

IPC分类号 : B01J27/06I,C01G29/00I,C02F1/72I,C02F101/30N,C02F101/34N





专利摘要
本发明公开了一种具有调节能带位置功能的氯氧化铋的制备方法及应用,属于环境保护和污染物降解技术领域。本发明利用乙醇、乙二胺、二甘醇和三甘醇中的任何多种种组成的混合溶剂来替代现有的乙醇作为反应溶剂,利用溶剂热反应制备得到价带下移的BiOCl材料。本发明的价带下移的BiOCl材料的光生h+的氧化能力更强,更有利于污染物的降解;此外这种强氧化能力的h+能够激活H2O2产生·O2‑,进一步提升光降解效率。光降解效果显示,在加入H2O2后,BiOCl对于罗丹明B、甲基橙以及苯酚的降解速率提升了3.2‑4.6倍。
权利要求
1.一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述方法包括以下步骤:将铋源和氯源分别溶于混合溶剂中,然后将氯源溶液逐滴加入到铋源溶液中并搅拌均匀,之后将所得悬浮液转移到水热反应釜中,并在120-220℃下加热16-24h,冷却后,洗涤,干燥,即可获得BiOCl,其中,所述混合溶剂为乙醇/二甘醇、乙醇/乙二胺、乙二胺/二甘醇、乙二胺/三甘醇或乙醇/乙二胺/二甘醇混合溶剂中的任一种。
2.根据权利要求1所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,当混合溶剂为乙醇/二甘醇、乙醇/乙二胺、乙二胺/二甘醇、乙二胺/三甘醇时,两种溶剂的体积比为2:1~1:2。
3.根据权利要求1或2所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述铋源和氯源的比例不限。
4.根据权利要求1或2所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述铋源为Bi(NO
5.根据权利要求3所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述铋源为Bi(NO
6.根据权利要求1、2或5任一所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述铋源溶液中以铋元素的浓度计,其浓度为0.02~0.04mol/L,所述氯源溶液中以氯元素计,其浓度为0.02~0.04mol/L。
7.根据权利要求3所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述铋源溶液中以铋元素的浓度计,其浓度为0.02~0.04mol/L,所述氯源溶液中以氯元素计,其浓度为0.02~0.04mol/L。
8.根据权利要求4所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述铋源溶液中以铋元素的浓度计,其浓度为0.02~0.04mol/L,所述氯源溶液中以氯元素计,其浓度为0.02~0.04mol/L。
9.根据权利要求1、2、5或7~8任一所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述干燥为将所得样品置于真空干燥箱中在真空条件下60-80℃干燥8-12h。
10.根据权利要求3所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述干燥为将所得样品置于真空干燥箱中在真空条件下60-80℃干燥8-12h。
11.根据权利要求4所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述干燥为将所得样品置于真空干燥箱中在真空条件下60-80℃干燥8-12h。
12.根据权利要求6所述的一种具有调节能带位置功能的氯氧化铋的制备方法,其特征在于,所述干燥为将所得样品置于真空干燥箱中在真空条件下60-80℃干燥8-12h。
13.权利要求1~12任一所述的一种具有调节能带位置功能的氯氧化铋的制备方法制备得到的BiOCl材料。
14.权利要求13所述BiOCl材料在光降解有机物中的应用,其特征在于,所述应用为:将BiOCl材料加入到有机污染物溶液中后,再加入H
15.根据权利要求14所述的应用,其特征在于,所述有机污染物包括罗丹明B、甲基橙或苯酚。
说明书
技术领域
本发明涉及一种具有调节能带位置功能的氯氧化铋的制备方法及应用,属于环境保护和污染物降解技术领域。
背景技术
近几十年来,在经济和工业化加速发展的同时,环境问题愈发严重,悄无生息的影响着我们赖以生存的家园,极大地降低了人们的生活质量。造纸、石油化工、染料、纺织等行业排放出大量的工业废水,严重污染了我们宝贵的淡水资源。因此,亟待开发出一种能够有效降解这些有机污染物的方式来解决日趋严重的环境问题。
目前,针对污染物降解主要采用Fenton法,臭氧氧化法、电化学氧化和光催化氧化等方法。相较于其它方法,光催化氧化去除有机污染物有如下优势:1、能够利用取之不尽,用之不竭的光能来激发光催化剂,成本较低;2、光催化材料普遍展现出的循环使用性进一步增加了其实际应用的可能性。
光催化材料用于降解有机污染物的一般作用机理为:当照射光的能量大于或等于半导体禁带宽度,在半导体内部产生电子-空穴对,随后电子(e
过氧化氢(H2O2)是一种强氧化剂,在环境保护和污染物降解领域有着较强的应用前景。如在Fenton降解中,H2O2能被体系中的Fe
发明内容
为了解决上述问题,本发明以调节能带位置和活性氧物种的产生为出发点,提供一种新型的氯氧化铋光催化剂的制备方法,来降解有机污染物分子。本发明通过选用特定的溶剂使得制备得到的BiOCl具有更正的价带位置(价带下移),从而使其光生h
具体的,首先,本发明的技术方案之一是提供一种具有调节能带位置功能的氯氧化铋的制备方法,所述方法包括以下步骤:将铋源和氯源分别溶于混合溶剂中,然后将氯源溶液逐滴加入到铋源溶液中并搅拌均匀,之后将所得悬浮液转移到至水热反应釜中,并在120-220℃下加热16-24h,待冷却后,洗涤,干燥,即可获得BiOCl,其中,所述混合溶剂为乙醇、乙二胺、二甘醇和三甘醇中的两种或多种组成的混合溶剂。
在本发明的一种实施方式中,当混合溶剂由乙醇、乙二胺、二甘醇和三甘醇中的任何两种组成时,两种溶剂的体积比优选为2:1~1:2。
在本发明的一种实施方式中,当混合溶剂为乙醇、乙二胺、二甘醇和三甘醇中的两种以上的溶剂组成时,其溶剂的混合比例不限。
在本发明的一种实施方式中,所述铋源和氯源的比例不限,优选铋元素和氯元素等摩尔量。
在本发明的一种实施方式中,所述铋源优选为Bi(NO3)3·5H2O或柠檬酸铋,更优选为Bi(NO3)3·5H2O,所述氯源优选为KCl、NaCl或十六烷基三甲基氯化铵中的一种或几种。
在本发明的一种实施方式中,所述铋源溶液中以铋元素的浓度计,其浓度为0.02~0.04mol/L,所述氯源溶液中以氯元素计,其浓度为0.02~0.04mol/L。
在本发明的一种实施方式中,所述搅拌的速率为200-600r/min,搅拌时间为1~3h。
在本发明的一种实施方式中,所述洗涤为用去离子水和乙醇交替洗涤3~7次。
在本发明的一种实施方式中,所述干燥为将所得样品置于真空干燥箱中在真空条件下60-80℃干燥8-12h。
在本发明的一种实施方式中,所述调节能带位置是指调节氯氧化铋的价带位置,价带位置的测定是通过X射线光电子仪测定。
其次,本发明提供了上述制备方法制备得到的BiOCl材料。
再者,本发明提供了上述BiOCl材料在光降解有机物中的应用。
在本发明的一种实施方式中,所述应用为:将BiOCl材料加入到有机污染物溶液中后,再加入H2O2后,在无光的情况下待吸附吸附平衡后,再在可见光下进行光降解。
在本发明的一种实施方式中,所述有机污染物包括但不限于罗丹明B、甲基橙和苯酚等。
在本发明的一种实施方式中,在降解有机污染物的过程中,在可见光的照射下,会产生活性氧物种,所述活性氧物种是指单线态氧(
在本发明的一种实施方式中,所述H2O2的加入量不限。
在本发明的一种实施方式中,所述H2O2一般以H2O2溶液的形式加入,加入量优选为:BiOCl和H2O2的摩尔比为1:2~10。
本发明取得的有益技术效果:
本发明通过反应溶剂的改性使得能够产生一种价带下移的BiOCl材料,且通过调节本发明的制备方法能够制备得到价带下移位置不同的BiOCl材料。本发明制备得到的BiOCl材料具有更正的价带位置(即价带下移),因此其光生h
附图说明
图1:实施例1中的BiOCl-1和对比例1中BiOCl-8的X射线光电子能谱-价带谱图(VB-XPS)。
图2:实施例1中BiOCl-1和对比例1中BiOCl-8在光催化过程中产生的·O2
图3:实施例1中BiOCl-1和对比例1中BiOCl-8在光催化过程中的电子自旋共振图谱(ESR)。
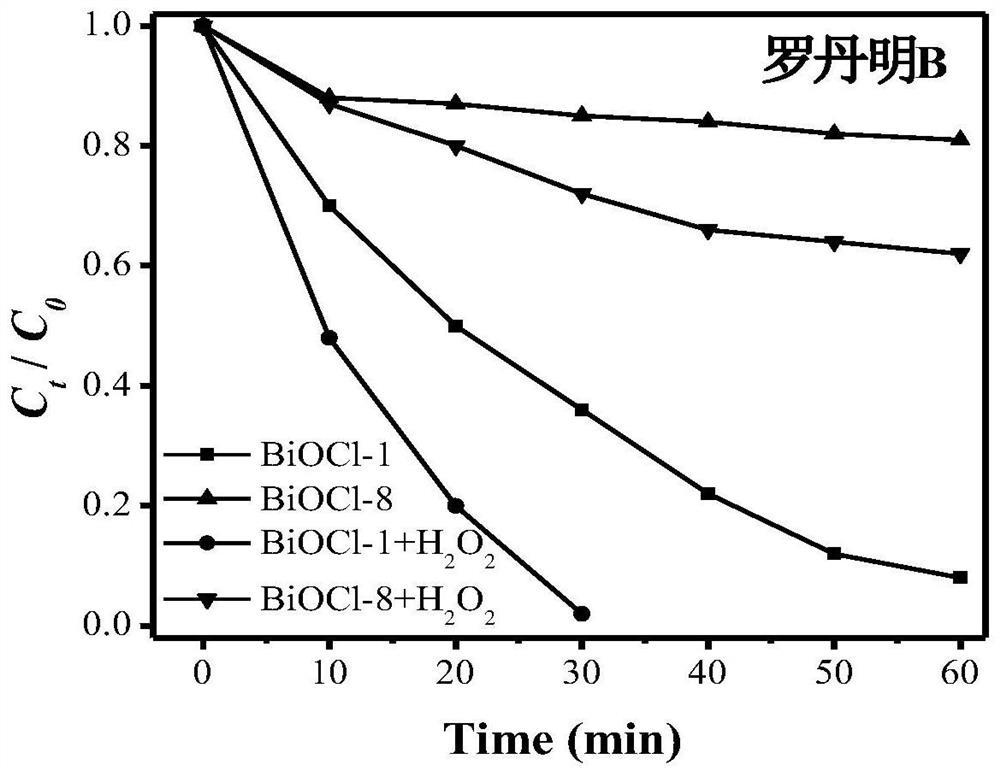
图4:实施例1中制备的BiOCl-1和对比例1中BiOCl-8样品对于罗丹明B的降解曲线。
图5:实施例1中制备的BiOCl-1和对比例1中BiOCl-8样品对于甲基橙的降解曲线。
图6:实施例1中制备的BiOCl-1和对比例1中BiOCl-8样品对于苯酚的降解曲线。
具体实施方式
下面实施例可以使本领域技术人员全面的理解本发明,但不以任何方式限制本发明。
光催化材料降解有机污染物:分别称量50mg制备的光催化材料加入到100mL浓度为10mg/L的罗丹明B、甲基橙和苯酚三种溶液中,并向得到的三种悬浮液中加入1mmol H2O2(市售30%的H2O2水溶液),黑暗中搅拌30min,然后在可见光(150W氙灯,λ>400nm)下进行光催化降解实验,光降解时间持续30-120min,每隔一段时间取3mL液体,离心后将上清液用紫外可见分光光度计或高效液相色谱仪测量以计算降解效果。
作为对比,光催化材料降解有机污染物的实验过程中不添加H2O2,其余步骤和条件和前述一致,测定其降解效果。
罗丹明B、甲基橙的降解效果用去除率表示,去除率的计算方法为:removal%=(C0-Ct)/C0×100%=(A0-At)/A0×100%,其中A0表示初始溶液的吸光度,At表示t时刻的吸光度,C0和Ct分别表示:初始溶液和t时刻溶液的浓度。
苯酚的降解效果用降解率表示,removal%=(C0-Ct)/C0×100%=(A0-At)/A0×100%,其中A0表示初始苯酚溶液的峰面积,At表示t时刻的峰面积,C0和Ct分别表示:初始溶液和t时刻溶液的浓度。
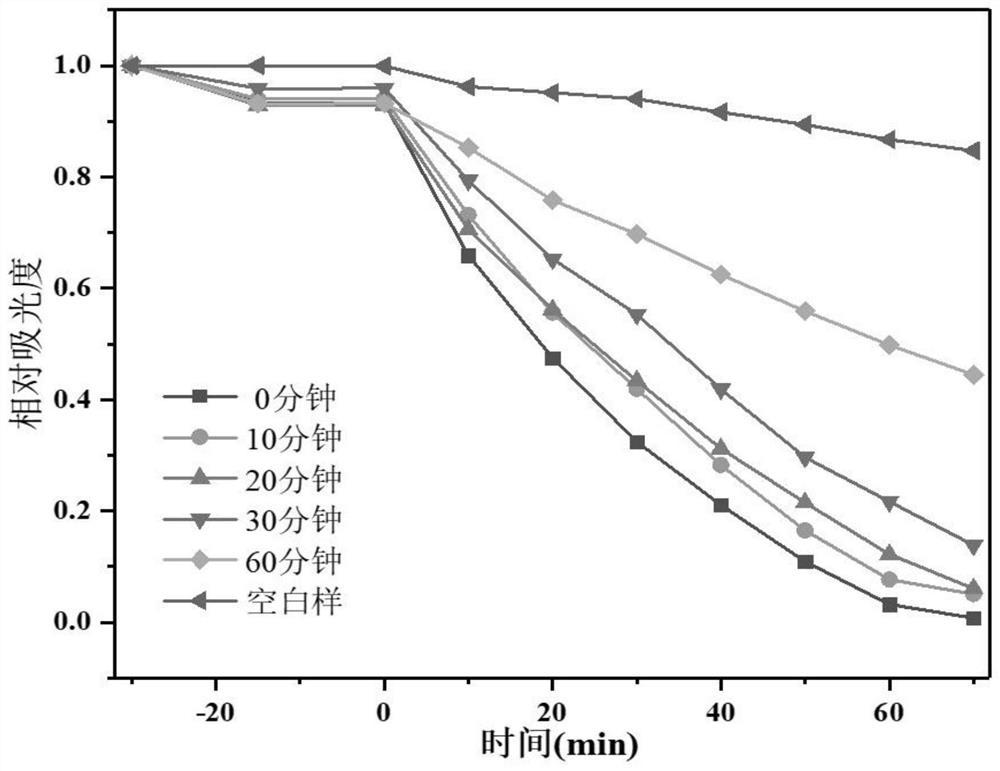
单线态氧和·O2
BiOCl的价带的测量:利用X射线光电子能谱仪测量。
实施例1
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙醇/二甘醇混合溶剂中(V乙醇:V二甘醇=1:1),得到浓度为10g/L悬浮液,在室温下超声1h;而后,同样地,称量KCl溶于40ml乙醇/二甘醇混合溶剂中(V乙醇:V二甘醇=1:1),得到浓度为1.5g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在400r/min下搅拌1h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在160℃下加热16小时,冷却至室温后,用去离子水和乙醇交替洗涤7次后,获得BiOCl样品,标记为BiOCl-1。
利用前述方法采用本实施例制备得到的BiOCl-1光降解有机污染物,降解方法如前面所述。光降解结果显示,未加H2O2时,60min后BiOCl-1对罗丹明B的降解率为92%,120min对甲基橙和苯酚降解率分别为50%和44%。当光降解过程中加入1mmol H2O2时,30min后BiOCl-1对罗丹明B的降解率即可为100%,120min对甲基橙和苯酚降解率分别为79%和80%。
特别地,相较于以乙醇为溶剂制备得到的BiOCl-8(对比例1),本实施例中通过乙醇/二甘醇改性合成得到的BiOCl-1具有更正的价带位置,经X射线光电子能谱仪测量材料的X射线光电子能谱-价带谱图,结果见图1,所得BiOCl-1价带下移了0.17eV,因此其光生h
图2为实施例1中BiOCl-1和对比例1中BiOCl-8在光催化过程中产生的·O2
图3为实施例1中BiOCl-1和对比例1中BiOCl-8在光催化过程中的电子自旋共振图谱(ESR),结果表明,相较于BiOCl-8,所得BiOCl-1以及添加H2O2后光催化体系中有着明显的
图4~6分别为实施例1中制备的BiOCl-1和对比例1中BiOCl-8样品对于罗丹明B、甲基橙和苯酚的降解曲线,可见,本发明的材料本身即具有较好的光催化性能,且在催化过程中添加了添加H2O2后,其催化活性大大提高。
实施例2
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙醇/二甘醇混合溶剂中(V乙醇:V二甘醇=1:2),得到浓度为10g/L悬浮液,在室温下超声2h;而后,同样地,称量KCl溶于40ml乙醇/二甘醇混合溶剂中(V乙醇:V二甘醇=1:2),得到浓度为1.5g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在600r/min下搅拌2h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在200℃下加热18小时,冷却至室温后,用去离子水和无水乙醇交替洗涤3次后,获得BiOCl样品,标记为BiOCl-2。
利用前述方法采用本实施例制备得到的BiOCl-2光降解有机污染物,光降解结果显示,未加H2O2时,60min后BiOCl-2对罗丹明B的降解率为87%,120min对甲基橙和苯酚降解率分别为46%和40%;当加入1mmol H2O2时,30min后BiOCl-2对罗丹明B的降解率达93%,120min对甲基橙和苯酚降解率分别为76%和74%。
特别地,相较于以乙醇为溶剂制备得到的BiOCl-8(对比例1),本实施例中所得BiOCl-2价带下移了0.32eV。
实施例3
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙二胺/二甘醇混合溶剂中(V乙二胺:V二甘醇=1:1),得到浓度为20g/L悬浮液,在室温下超声0.5h;而后,同样地,称量KCl溶于40ml乙二胺/二甘醇混合溶剂中(V乙二胺:V二甘醇=1:1),得到浓度为3g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在200r/min下搅拌1h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在160℃下加热16小时,冷却至室温后,用去离子水和乙醇交替洗涤5次后,获得BiOCl样品,标记为BiOCl-3。
利用前述方法采用本实施例制备得到的BiOCl-3光降解有机污染物。光降解结果显示,未加H2O2时,60min后BiOCl-3对罗丹明B的降解率为90%,120min对甲基橙和苯酚降解率分别为48%和44%,当加入1mmol H2O2时,30min后BiOCl-3对罗丹明B的降解率为92%,120min对甲基橙和苯酚降解率分别为70%和72%。
特别地,相较于以乙醇为溶剂制备得到的BiOCl-8(对比例1),本实施例中所得BiOCl-3价带下移了0.22eV。
实施例4
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙二胺/二甘醇混合溶剂中(V乙二胺:V二甘醇=1:2),得到浓度为15g/L悬浮液,在室温下超声1h;而后,同样地,称量KCl溶于40ml乙二胺/二甘醇混合溶剂中(V乙二胺:V二甘醇=1:2),得到浓度为2.25g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在400r/min下搅拌2h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在120℃下加热20小时,冷却至室温后,用去离子水和乙醇交替洗涤7次后,获得BiOCl样品,标记为BiOCl-4。
利用前述方法采用本实施例制备得到的BiOCl-4光降解有机污染物。光降解结果显示,未加H2O2时,60min后BiOCl-4对罗丹明B的降解率为88%,120min对甲基橙和苯酚降解率分别为45%和42%,当加入1mmol H2O2时,30min后BiOCl-4对罗丹明B的降解率为90%,120min对甲基橙和苯酚降解率分别为73%和71%。
特别地,相较于以乙醇为溶剂制备得到的BiOCl-8(对比例1),本实施例中所得BiOCl-4价带下移了0.36eV。
实施例5
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙醇/乙二胺混合溶剂中(V乙醇:V乙二胺=1:2),得到浓度为20g/L悬浮液,在室温下超声0.5h;而后,同样地,称量KCl溶于40ml乙醇/乙二胺混合溶剂中(V乙醇:V乙二胺=1:2),得到浓度为3g/L悬浮液,在室温下超声0.5h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在400r/min下搅拌1h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在120℃下加热20小时,冷却至室温后,用去离子水和乙醇交替洗涤5次后,获得BiOCl样品,标记为BiOCl-5。
利用前述方法采用本实施例制备得到的BiOCl-5光降解有机污染物。光降解结果显示,未加H2O2时,60min后BiOCl-1对罗丹明B的降解率为92%,120min对甲基橙和苯酚降解率分别为40%和44%,当加入1mmol H2O2时,30min后BiOCl-1对罗丹明B的降解率为96%,120min对甲基橙和苯酚降解率分别为78%和76%。
特别地,相较于以乙醇为溶剂制备得到的BiOCl-8(对比例1),本实施例中所得BiOCl-5价带下移了0.4eV。
实施例6
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙醇/二甘醇混合溶剂中(V乙醇:V三甘醇=1:1),得到浓度为10g/L悬浮液,在室温下超声1h;而后,同样地,称量KCl溶于40ml乙醇/三甘醇混合溶剂中(V乙醇:V三甘醇=1:1),得到浓度为1.5g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在400r/min下搅拌2h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在120℃下加热20小时,冷却至室温后,用去离子水和乙醇交替洗涤5次后,获得BiOCl样品,标记为BiOCl-6。
利用前述方法采用本实施例制备得到的BiOCl-6光降解有机污染物。光降解结果显示,未加H2O2时,60min后BiOCl-6对罗丹明B的降解率为90%,120min对甲基橙和苯酚降解率分别为40%和44%,当加入1mmol H2O2时,30min后BiOCl-6对罗丹明B的降解率为95%,120min对甲基橙和苯酚降解率分别为72%和74%。
特别地,相较于以乙醇为溶剂制备得到的BiOCl-8(对比例1),本实施例中所得BiOCl-6价带下移了0.31eV。
实施例7
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙二胺/三甘醇混合溶剂中(V乙二胺:V三甘醇=1:2),得到浓度为15g/L悬浮液,在室温下超声2h;而后,同样地,称量KCl溶于40ml乙二胺/三甘醇混合溶剂中(V乙二胺:V三甘醇=1:2),得到浓度为2.25g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在600r/min下搅拌2h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在180℃下加热18小时,冷却至室温后,用去离子水和无水乙醇交替洗涤3次后,获得BiOCl样品,标记为BiOCl-7。
利用前述方法采用本实施例制备得到的BiOCl-7光降解有机污染物。光降解结果显示,未加H2O2时,60min后BiOCl-7对罗丹明B的降解率为93%,120min对甲基橙和苯酚降解率分别为59%和46%,当加入1mmol H2O2时,30min后BiOCl-7对罗丹明B的降解率为98%,120min对甲基橙和苯酚降解率分别为80%和78%。
特别地,相较于以乙醇为溶剂制备得到的BiOCl-8(对比例1),本实施例中所得BiOCl-7价带下移了0.34eV。
实施例8
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙醇/乙二胺/二甘醇混合溶剂中(V乙醇:V乙二胺:V二甘醇=1:1:2),得到浓度为10g/L悬浮液,在室温下超声1h;而后,同样地,称量KCl溶于40ml乙醇/乙二胺/二甘醇混合溶剂中(V乙醇:V乙二胺:V二甘醇=1:1:2),得到浓度为1.5g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在400r/min下搅拌1h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在160℃下加热16小时,冷却至室温后,用去离子水和乙醇交替洗涤7次后,获得BiOCl样品。
利用前述方法采用本实施例制备得到的BiOCl光降解有机污染物。光降解结果显示,未加H2O2时,60min后BiOCl对罗丹明B的降解率为84%,120min对甲基橙和苯酚降解率分别为52%和48%,当加入1mmol H2O2时,30min后BiOCl对罗丹明B的降解率为92%,120min对甲基橙和苯酚降解率分别为78%和76%。
实施例9
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙醇/乙二胺/二甘醇混合溶剂中(V乙醇:V乙二胺:V二甘醇=10:1:1),得到浓度为10g/L悬浮液,在室温下超声1h;而后,同样地,称量NaCl溶于40ml乙醇/乙二胺/二甘醇混合溶剂中(V乙醇:V乙二胺:V二甘醇=10:1:1),得到浓度为1.5g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在400r/min下搅拌1h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在160℃下加热16小时,冷却至室温后,用去离子水和乙醇交替洗涤7次后,获得BiOCl样品。
利用前述方法采用本实施例制备得到的BiOCl光降解有机污染物。光降解结果显示,未加H2O2时,60min后BiOCl对罗丹明B的降解率为80%,120min对甲基橙和苯酚降解率分别为47%和44%,当加入1mmol H2O2时,30min后BiOCl对罗丹明B的降解率为88%,120min对甲基橙和苯酚降解率分别为75%和72%。
对比例1
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml乙醇中,得到浓度为10g/L悬浮液,在室温下超声1h;而后,同样地,称量KCl溶于40ml乙醇中,得到浓度为1.5g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在400r/min下搅拌1h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在160℃下加热16小时,冷却至室温后,用去离子水和乙醇交替洗涤7次后,获得BiOCl样品,标记为BiOCl-8。
利用前述方法采用制备得到的BiOCl-8光降解有机污染物。光降解结果显示,未加H2O2时,BiOCl-8在60min后对罗丹明B的降解率仅为20%。当加入1mmol H2O2时,BiOCl-8在60min后对罗丹明B的降解率也仅为38%。可见,使用乙醇作为溶剂制备得到的BiOCl无法大量激活H2O2产生·O2
对比例2
Fe-BiOCl的制备:称取0.485g Bi(NO3)3·5H2O溶于40ml 0.1M甘露醇溶液中,在200r/min下搅拌30min,随后向上述悬浮液中加入0.2g聚乙烯吡咯烷酮(PVP K30)并继续搅拌10min,然后30ml 0.1M FeCl2·4H2O加入到上述悬浮液中,然后0.0745g KCl加入到上述悬浮液,反应30min,随后转移至聚四氟乙烯反应釜中,在180℃下保持4h,自然冷却至室温后,所得样品经离心、干燥得Fe掺杂BiOCl纳米片,标记为Fe-BiOCl。
利用前述方法采用制备得到的Fe-BiOCl光降解罗丹明B,光降解结果显示,未加H2O2时,Fe-BiOCl在60min后对于罗丹明B降解率为80%。当加入1mmol H2O2时,Fe-BiOCl在60min后对于罗丹明B降解率为88%,略有提升。
对比例3
Ag/AgCl/BiOCl的制备:0.82g of Bi(NO3)3·5H2O和0.024g AgNO3溶于50ml去离子水中,搅拌30min,随后40ml 0.01M十六烷基三甲基氯化铵(CTAC)加入到上述悬浮液,在80℃下水浴反应3h,所得样品经离心、干燥得Ag/AgCl/BiOCl。
利用前述方法采用制备得到的Ag/AgCl/BiOCl光降解罗丹明B,光降解结果显示,未加H2O2时,Ag/AgCl/BiOCl在70min后对于罗丹明B降解率为82%,当加入1mmol H2O2时,Ag/AgCl/BiOCl在70min后对于罗丹明B降解率为90%,略有提升。
对比例4
Bi/BiOCl的制备:称取0.485g Bi(NO3)3·5H2O和0.0745g KCl分别溶于40ml去离子水中,超声溶解10min,随后将KCl溶液逐滴加入到Bi(NO3)3·5H2O悬浮液中,200r/min下搅拌30min,随后转移至聚四氟乙烯反应釜中,在160℃下保持24h,自然冷却至室温,经离心、干燥可得BiOCl纳米片。称取0.1g所得BiOCl加入到100ml 0.1g·L草酸铵中,在4W的紫外灯下照射30min,然后离心、干燥得到Bi沉积BiOCl材料,标记为Bi/BiOCl。
Bi/BiOCl光降解罗丹明B:称量50mg Fe-BiOCl加入到100mL浓度为10mg/L的罗丹B水溶液中,黑暗中搅拌30min,然后在紫外光下进行光催化降解实验,光降解时间持续80min,每隔一段时间取3ml液体,离心后将上清液用紫外可见分光光度计测量计算降解效果,结果表明Bi/BiOCl 80min对于罗丹明B降解率为75%。
对比例5
BiOCl的制备:称量Bi(NO3)3·5H2O溶于40ml去离子水中,得到浓度为10g/L悬浮液,在室温下超声1h;而后,同样地,称量KCl溶于40ml去离子水中,得到浓度为1.5g/L悬浮液,在室温下超声1h;随后将KCl悬浮液逐滴加入到Bi(NO3)3·5H2O悬浮液中,并在400r/min下搅拌1h。之后,将所得悬浮液转移到至聚四氟乙烯内衬的高压水热反应釜中,并在160℃下加热16小时,冷却至室温后,用去离子水洗涤3次后,获得BiOCl样品,标记为BiOCl-9。
利用前述方法采用制备得到的BiOCl-9光降解有机污染物。光降解结果显示,未加H2O2时,BiOCl-8在60min后对罗丹明B的降解率仅为16%。当加入1mmol H2O2时,BiOCl-9在60min后对罗丹明B的降解率也仅为34%。可见,以水作为溶剂制备的BiOCl对于罗丹明B降解能力很小。
对比例6
取1mmol H2O2(市售30%的H2O2水溶液)加入到100mL浓度为10mg/L的罗丹明B溶液中,黑暗中搅拌30min,然后在可见光(150W氙灯,λ>400nm)下进行光催化降解实验,光降解时间持续60min,每隔一段时间取3mL液体,将上清液用紫外可见分光光度计测量以计算降解效果。可以发现,60min后,H2O2对罗丹明B的降解率仅为2%左右,几乎无降解效果。
可见,本发明中通过调控价带位置使得具有更强氧化能力的光生h
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
一种具有调节能带位置功能的氯氧化铋的制备方法及应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。














动态评分
0.0