专利摘要
本发明提供了一种聚阴离子Na8H32[{MnIV24MnIII12O28(H2O)23}2(W24O120)2]·66H2O(记为Mn72W48)的制备方法,将Mn(CH3COO)2和KMnO4溶解在稀释的CH3COOH溶液中并持续搅拌获得sol.Mn溶液,然后向sol.Mn溶液中逐滴加入Na2WO4溶液,利用Na2WO4的碱性将sol.Mn溶液的pH升高至为5.5左右,获得sol.Mn‑W溶液。将sol.Mn‑W溶液加热冷却并过滤后通过蒸发获得。本发明的合成工艺使溶液中锰物质的浓度得到了极大地提高,对溶液中低核锰簇的质谱信号不存在监控盲区,进一步提高了合成的质量和效率。
权利要求
1.一种高核金属锰簇的制备方法,其特征是,对制备方法进行实时监测,对溶液中低核锰簇的质谱信号不存在监控盲区,能够检测Mn2(Ac)5-锰簇简称Mn2、Mn3(Ac)7-锰簇简称Mn3,Mn4O2(Ac)8-锰簇简称Mn4,HMn6O6(Ac)92+锰簇简称Mn6,K2H3Mn7O8(Ac)122+锰簇简称Mn7,KMn2W4O13(Ac)6-锰簇简称Mn2W4,Mn2W5O16(Ac)52-锰簇简称Mn2W5和Mn3W5O16(Ac)72-锰簇简称Mn3W5的存在;
具体步骤为:
步骤1,将Mn(CH3COO)2和KMnO4溶解在稀释的CH3COOH溶液中并持续搅拌1个小时以上形成[MnIII8MnIV4O12(CH3COO)16(H2O)4]溶液,简称为sol.Mn溶液;
步骤2,向步骤1获得的sol.Mn溶液中缓慢加入适量Na2WO4溶液,使得溶液pH值降至5.5,获得sol.Mn-W溶液;
步骤3,将sol.Mn-W溶液加热冷却并过滤,然后通过蒸发得到红黑色板状晶体,所述晶体为聚阴离子Na8H32[{MnIV24MnIII12O28(H2O)23}2(W24O120)2]·66H2O晶体,简称为Mn72W48晶体,所述聚阴离子Mn72W48晶体包含两个[MnIV24MnIII12O28(H2O)23W24O120]20-笼状核,简称Mn36W24笼状核;
所述实时监测过程为:
向所述sol.Mn中加入Na2WO4使Na2WO4的浓度逐渐增大到5×10-6mol/L的过程中进行质谱监测,显示出随着Na2WO4的浓度逐渐增大Mn2和Mn3簇的峰值强度也在逐渐增强,同时溶液中开始出现Mn12(Ac)18O12(HAc)(H2O)3W5O162-团簇简称Mn12W5,[MnIII8MnIV4O12(CH3COO)16(H2O)4]简称为Mn12作为溶液中的主要物种,浓度变化不大;继续向溶液中添加Na2WO4,当Na2WO4的浓度达到7×10-5mol/L时,Mn2W4,Mn2W5和Mn3W5开始出现,此时Mn12及Mn2,Mn3,Mn12W5的浓度均有不同程度的降低,并且原始Mn12簇的峰强度急剧下降,Mn2和Mn3在经过短暂的强度减小后开始增加并在Na2WO4的浓度达到1×10-4M时,峰强度达到最大值;
继续加入Na2WO4,Mn2,Mn3和Mn12的含量均开始逐渐下降,而Mn12W5,Mn2W4,Mn2W5和Mn3W5的所有峰强度都逐渐增加,同时检测到H2W10O322-簇峰强度增加明显;
当Na2WO4的浓度达到1×10-3mol/L时,Mn-W物种和H2W10O322-簇的峰强度最大;
继续向溶液中加入Na2WO4,Mn-W物种和H2W10O322-簇的所有峰强度开始随着Mn4,Mn6和Mn7物种的形成而降低,W6O19(NaAc)52-簇的强度也随之增强,之后,当Na2WO4浓度增长到1×10-3mol/L时Mn12簇会再次出现。
2.根据权利要求1所述的高核金属锰簇的制备方法,其特征是:所述Mn72W48晶体中两个Mn36W24笼状核,通过两个Mn-O-W键连接;
每个Mn36W24基团含有四个Mn6W6平面和六个Mn2团簇;
在Mn6W6基团中,三个W2同多金属氧酸盐单元分别位于平面三角形MnIV6单元的边缘,形成六边形平面结构;
四个Mn6W6六边形通过W-O-Mn键由六个Mn2簇连接,构成了Mn36W24团簇。
说明书
技术领域
本发明属于高核金属锰簇制备领域,具体地涉及一种聚阴离子Mn72W48的制备方法。
背景技术
高核锰簇在催化、磁学以及医学等领域具有重大的应用价值,因而得到了广泛的关注。合成高核锰簇的方法通常是使用有机配体将低核锰簇连接起来,但是这种方法的弊端在于需要使用有毒易挥发且高度易燃的有机溶剂。而作为无机多齿配体的多金属氧酸盐(POMs),具有结构易变可控的特点,同时可以在水相中进行反应合成高核的金属簇,从而使整个反应更加经济环保。但是多金属氧酸盐(POMs)作为无机多齿配体进行簇连,对溶液的酸碱性和制备的工艺流程要求极为苛刻,对工艺流程进行实时监控成为本领域的迫切需要。
包含两个[MnIV24MnIII12O28(H2O)23W24O120]20-(Mn36W24)笼状核的Na8H32[{MnIV24MnIII12O28(H2O)23}2(W24O120)2]·66H2O(简称为Mn72W48)聚阴离子,因其潜在的高催化活性得到了广泛的关注。目前合成Mn72W48聚阴离的方案较由于受到合成方法限制,无法通过高分辨质谱等手段,具体监控在合成过程中的起始原料是如何变为最终Mn72W48的,即不能实现实时监控。现有的制备Mn72W48的方法是通过CH3COOH,Mn(CH3COO)2,KMnO4和Na2WO4在水溶液中反应合成的,具体为:向浓度较高的Na2WO4溶液中添加[MnIII8MnIV4O12(CH3COO)16(H2O)4]溶液(以下简以sol.Mn表示),利用[MnIII8MnIV4O12(CH3COO)16(H2O)4]溶液的酸性将体系调节pH值到5.5来合成Mn72W48。在合成过程中需要对原始浓度的溶液进行质谱分析监控,以避免高浓度碱性Na2WO4溶液对溶液中其他物质造成影响。通过计算可知,在反应原料均匀混合后,钨酸盐和Mn物种的物质的量之比约为45∶1(溶液中存在1g钨酸钠和0.045g Mn12),锰物种的高分辨质谱信号大部分被高浓度的钨酸盐所掩盖而无法检测,从而限制了人们对该化合物合成进程的监控,制约了对于该合成过程中存在的其他低核锰簇变化的研究。
发明内容
为了解决现有Mn72W48聚阴离子合成工艺中存在的问题,我们提出了一个新的合成工艺,同样得到了聚阴离子Mn72W48,并且使溶液中锰物质的浓度得到了极大地提高,能够对合成工艺进行实时监测,对溶液中低核锰簇的质谱信号不存在监控盲区,进一步提高了合成的质量和效率,以及在检测过程中检测到了Mn2(Ac)5-(Mn2),Mn3(Ac)7-(Mn3),Mn4O2(Ac)8-(Mn4),HMn6O6(Ac)92+(Mn6),K2H3Mn7O8(Ac)122+(Mn7),KMn2W4O13(Ac)6-(Mn2W4),Mn2W5O16(Ac)52-(Mn2W5)和Mn3W5O16(Ac)72-(Mn3W5)等锰簇的存在,并且通过监测这些锰簇的浓度变化,进一步完善了聚阴离子Mn72W48的合成机理。
本发明的技术解决方案是:
将Mn(CH3COO)2和KMnO4溶解在稀释的CH3COOH溶液中并持续搅拌1个小时以上,获得sol.Mn溶液,然后向sol.Mn溶液中缓慢加入适量Na2WO4溶液,使得溶液pH值降至5.5左右,获得sol.Mn-W溶液,将sol.Mn-W溶液加热冷却并过滤后通过蒸发得到了红黑色的板状晶体,所得晶体即为聚阴离子Mn72W48.
具体有以下步骤制备:
步骤1,[MnIII8MnIV4O12(CH3COO)16(H2O)4]溶液的合成。
将Mn(CH3COO)2·4H2O溶于体积分数为60%的CH3COOH溶液中,完全溶解后,向溶液中一次性加入适量KMnO4,继续搅拌至少一个小时,获得[MnIII8MnIV4O12(CH3COO)16(H2O)4]溶液,简写为sol.Mn溶液。
步骤2,Na8H32[{MnIV24MnIII12O28(H2O)23}2(W24O120)2]·66H2O(Mn72W48)的合成
用适量体积分数为10%的CH3COOH溶液对适量步骤1制备获得的sol.Mn溶液进行稀释,逐滴加入Na2WO4溶液,使溶液pH值升高到5.5左右,获得sol.Mn-W溶液。随后将深橙色/棕色混合溶液加热到90℃,保持数小时后,随后冷却至室温并过滤出无色块状晶体十二钨酸盐([H2W12O42]10-),静置并挥发滤液,获得Na8H32[{MnIV24MnIII12O28(H2O)23}2(W24O120)2]·66H2O(Mn72W48)晶体。
在整个合成过程中,反应溶液可以随时取出用于质谱检测分析,从而验证溶液中所存在的主要物种。IR(KBr粉末,υ/cm-1):3424(br/m),1629(m),943(w),826(m),682(s)。Mn72W48晶体主要晶体学数据:三斜晶系,P-1空间群, α=93.914(1)°,β=96.564(1)°,γ=104.502(1)°,2=1,
本发明的有益效果是:
本发明通过对合成方法进行重新设计,将Mn(CH3COO)2和KMnO4溶解在稀释的CH3COOH溶液中并持续搅拌获得sol.Mn溶液,然后向sol.Mn溶液中逐滴Na2WO4溶液,使得溶液pH升高到5.5,获得sol.Mn-W溶液,将sol.Mn-W溶液加热冷却并过滤后通过蒸发得到了红黑色的板状晶体。将所得红黑色的板状晶体即为Mn72W48。在整个合成过程中,反应溶液可随时取出进行质谱测试,验证反应溶液中存在的主要物种,实现了对工艺流程进行实时监控。(Mn72W48)红外测试和X-射线单晶衍射测试(Mn72W48)结果显示,红外光谱和晶胞参数与本领域现有方法制备出的晶体完全吻合。
本发明的合成工艺使溶液中锰物质的浓度得到了极大地提高,对溶液中低核锰簇的质谱信号不存在监控盲区,进一步提高了合成的质量和效率,以及在检测过程中检测到了Mn2(Ac)5-(Mn2),Mn3(Ac)7-(Mn3),Mn4O2(Ac)8-(Mn4),HMn6O6(Ac)92+(Mn6),K2H3Mn7O8(Ac)122+(Mn7),KMn2W4O13(Ac)6-(Mn2W4),Mn2W5O16(Ac)52-(Mn2W5)和Mn3W5O16(Ac)72-(Mn3W5)等锰簇的存在,并且通过监测这些锰簇的浓度变化,进一步完善了聚阴离子Mn72W48的合成机理。
附图说明
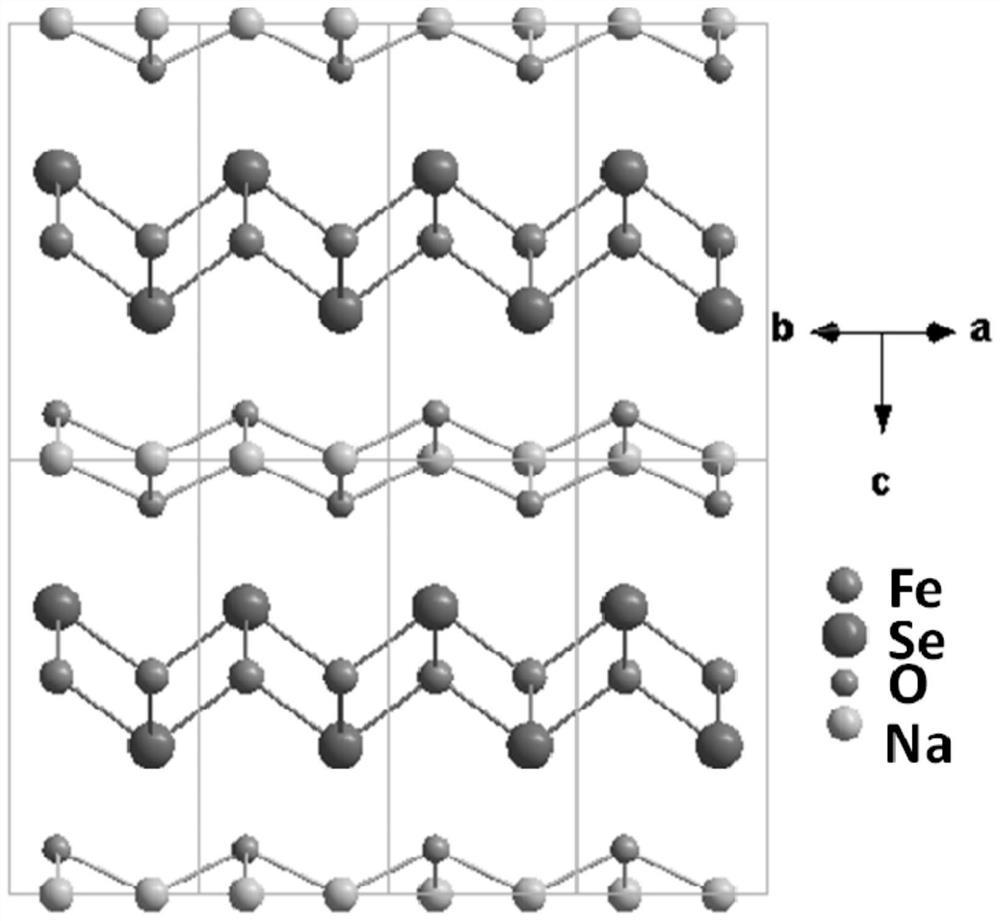
附图1是聚阴离子Mn72W48的结构如图1所示。每个Mn36W24笼的结构单元包括4个MnIV6基团,12个W2基团和6个MnIII2)基团。MnIV6及W2组成的六边形平面结构。
附图2是在负离子模式下,sol.Mn-W中部分物种的实验及模拟高分辨质谱图。
附图3是sol.Mn-W中各物种随Na2WO4浓度变化的高分辨质谱峰强度。
附图4是Mn72W48的聚合机理示意图。
具体实施方式
下面通过实施进一步详细描述本发明,但这并非是对本发明的限制,根据本发明的基本思想,可以做出各种修改和改进,但只要不脱离本发明的基本思想,均在发明的范围之内。
[实施例]
一、Mn72W48聚阴离子的制备方法。
1.1、[MnIII8MnIV4O12(CH3COO)16(H2O)4]溶液的合成
将1.00g 4.08mmol的Mn(CH3COO)2·4H2O溶于10mL体积分数为60%的CH3COOH溶液中,完全溶解后,向溶液中加入0.25g 1.58mmol KMnO4粉末,继续搅拌至少一个小时,获得sol.Mn溶液
1.2 Na8H32[{MnIV24MnIII12O28(H2O)23}2(W24O120)2]·66H2O(Mn72W48)的合成
用30mL体积分数对10%的CH3COOH溶液将0.365mL sol.Mn溶液进行稀释,并向sol.Mn溶液中逐滴加入1mol/L Na2WO4溶液,使sol.Mn溶液的pH升高至5.5左右,最终溶液记为sol.Mn-W溶液。然后将深橙色/棕色sol.Mn-W溶液热到90℃,并保持1小时,随后冷却至室温并过滤。静止24小时后将无色块状晶体十二钨酸盐([H2W12O42]10-)滤出。继续静止一周后,在滤液中获得呈红黑色的板状晶体(Mn72W48)。Mn72W48表征:IR(KBr粉末,υ/cm-1):3424(br/m),1629(m),943(w),826(m),682(s)。Mn72W48主要晶体学数据:三斜晶系,P-1空间群, α=93.914(1)°,β=96.564(1)°,γ=104.502(1)°,Z=1,
二、Mn72W48聚阴离子的结构。
制备出的聚阴离子Mn72W48的结构如图1所示,Mn72W48聚阴离子包含两个[MnIV24MnIII12O28(H2O)23W24O120]20-(Mn36W24)笼状核,它们通过两个Mn-O-W键连接。每个Mn36W24基团含有四个Mn6W6平面和六个Mn2团簇:在Mn6W6基团中,三个W2同多金属氧酸盐单元分别位于平面三角形MnIV6单元的边缘,形成六边形平面结构。这样的四个Mn6W6六边形通过W-O-Mn键由六个Mn2簇连接,构成了Mn36W24团簇。
三、Mn72W48聚阴离子合成过程讨论
Na8H32[{MnIV24MnIII12O28(H2O)23}2(W24O120)2]·66H2O(Mn72W48)是通过CH3COOH,Mn(CH3COO)2,KMnO4和Na2WO4在水溶液中反应合成的。将Mn(CH3COO)2和KMnO4溶解在稀释的CH3COOH溶液中并持续搅拌获得sol.Mn溶液,然后向sol.Mn溶液中逐滴加入Na2WO4溶液将pH设置为5.5,获得sol.Mn-W溶液,将sol.Mn-W溶液加热冷却并过滤后通过蒸发得到了红黑色的板状晶体。将所得红黑色的板状晶体即为Mn72W48。对Mn72W48晶体进行红外测试和X-射线单晶衍射测试,结果显示,红外光谱和晶胞参数与本领域现有工艺制备出的晶体完全吻合。本发明的合成工艺使溶液中锰物质的浓度得到了极大地提高,对溶液中低核锰簇的质谱信号不存在监控盲区。
在合成中还需要注意到Na2WO4在Mn12簇的重排中起的重要作用。如果用Na2MoO4取代Na2WO4,则唯一的最终产物仅为Waugh型Na6MnMo9O32·xH2O。如果用Li2WO4,K2WO4或(NH4)2WO4代替Na2WO4,也不能得到Mn72W48。另外,Mn6W6中间体是比较稳定的。若在合成过程中首先用NaOH与sol.Mn混合,Mn12簇将被破坏,再加入Na2WO4并不能得到Mn6W6中间体。然而,如果向sol.Mn-W中加入NaOH溶液,并不会影响Mn72W48的合成。
四、Mn72W48聚阴离子合成过程质谱监测
在合成过程中保持ESI-MS测试样品的浓度与合成过程中相同,以避免浓度对溶液中物质的影响。将sol.Mn-W的所有ESI-MS谱图与HAc溶液中的Na2WO4的相应谱图进行比较,从而排除CH3COONa或其他物质的干扰。图2列出了sol.Mn-W中检测到的部分锰簇信号。
向sol.Mn中加入Na2WO4使Na2WO4的浓度逐渐增大到5×10-6mol/L(单位简以M代表)的过程中的质谱监测结果,如图3所示,随着Na2WO4的浓度逐渐增大Mn2和Mn3簇的峰值强度也在逐渐增强,同时我们观察到溶液中开始出现Mn12(Ac)18O12(HAc)(H2O)3W5O162-(Mn12W5)团簇,而Mn12作为溶液中的主要物种,浓度变化不大。随着继续向溶液中添加Na2WO4,当Na2WO4的浓度达到7×10-5M时,更多的Mn-W物种(Mn2W4,Mn2W5和Mn3W5)开始出现,此时Mn12及Mn2,Mn3,Mn12W5的浓度均有不同程度的降低,并且原始Mn12簇的峰强度急剧下降,Mn2和Mn3在经过短暂的强度减小后开始增加并在Na2WO4的浓度达到1×10-4M时,峰强度达到最大值。此后,随着Na2WO4的加入,Mn2,Mn3和Mn12的含量均开始逐渐下降,而Mn-W物种(Mn12W5,Mn2W4,Mn2W5和Mn3W5)的所有峰强度都逐渐增加,同时检测到了H2W10O322-(W10)峰强度增加明显。当Na2WO4的浓度达到1×10-3M时,观察到Mn-W物种和W10簇的最大峰强度。随着继续向溶液中加入Na2WO4,Mn-W物种和W10簇的所有峰强度开始随着Mn4,Mn6和Mn7物种的形成而降低,W6O19(NaAc)52-(W6)簇的强度也随之增强。经过如此复杂的物种转化后,当Na2WO4浓度增长到1×10-3M时Mn12簇会再次出现。
五、Mn72W48聚阴离子合成机理
根据Mn72W48聚阴离子合成过程质谱监测结果,Mn72W48的合成机理如图4所示,向Mn12溶液中加入Na2WO4时,一开始溶液中低核锰团簇(Mn1,Mn2,Mn3)的浓度会随着Na2WO4浓度的增大而增大,说明Na2WO4可以将Mn12团簇解离成低核锰簇。当继续加入Na2WO4,溶液中同多钨酸盐的含量增加并迅速同这些低核锰簇结合在一起,不断地将它们从有机乙酸配位的锰团簇重新排列成无机多金属氧酸盐配位的锰钨簇(Mn2W4,Mn2W5,Mn3W5)。Na2WO4浓度进一步提高生成大量同多钨酸盐(W1~W6)的同时,锰钨簇再次解离成Mn4,Mn6,Mn7,最终锰簇与同多钨酸盐结合变成相对稳定的Mn6W6和MnW6离子。与此同时,溶液中还存在少量的低核锰簇。其中,Mn2簇与Mn6W6结合,生成了Mn72W48。
由于溶液中存在大量的钨物种,因此仲钨酸盐W12会先于Mn72W48从溶液中结晶出来。
一种高核金属锰簇的制备方法专利购买费用说明

Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。

















动态评分
0.0