

IPC分类号 : B01J23/58,C01G55/00,C25B1/04,C25B11/06









专利摘要
一类纯相铱酸锶类催化剂、制备方法及其在高效电催化裂解酸性水产氧方面的应用,属于无机功能材料领域。是将铱源、有机多元醇、锶源、有机多元酸和水通过不同比例混合加热蒸干,然后将蒸干的反应物煅烧一段时间,再将煅烧得到的产物用0.5~2mol/L的盐酸、硫酸、高氯酸等浸泡3~10h,即可得到不同相的铱酸锶。其中所得的单斜相铱酸锶SrIrO3结晶度高,呈规则的六边形片,可代替传统的酸性水氧化催化剂二氧化铱,在一定程度上降低了贵金属铱的用量。其电催化水裂解析氧电流密度达到10mA/cm2时,仅需过电势248mV,在目前报道过的水裂解析氧催化剂中本征活性最高,远远好于目前工业所用贵金属催化剂,并且性能稳定不衰减,具有广阔的应用前景。
权利要求
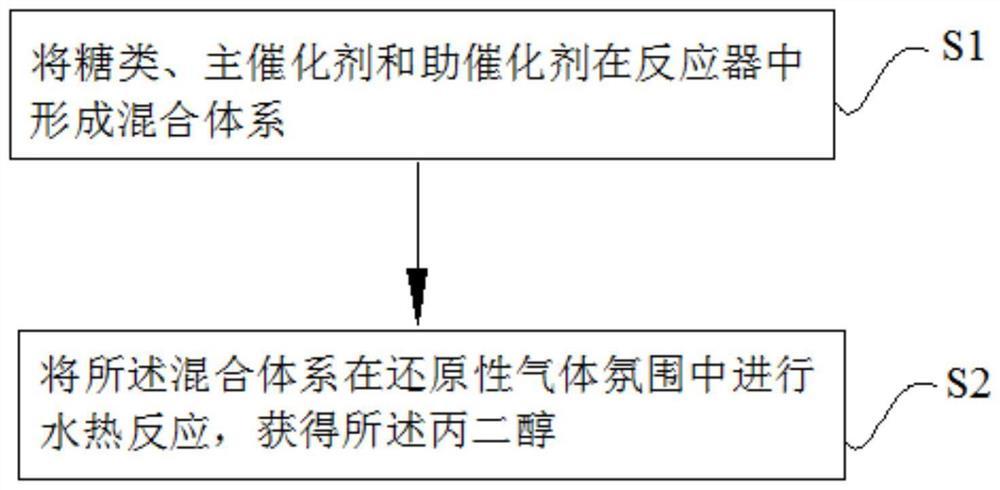
1.一类铱酸锶催化剂的制备方法,其步骤如下:
(1)混合溶液的配制:称取n
(2)干燥和煅烧:将步骤(1)得到的混合溶液在120~200℃环境中干燥5~10h,将干燥后的固态样品研磨成粉末后以0.5~5℃/min的升温速率升温,然后在120~220℃、230~320℃、330~520℃和620~720℃下分别加热1~10h、1~10h、2~4h和1~10h,自然冷却至室温得到黑色粉末;
(3)酸处理:将步骤(2)最终得到的黑色粉末用0.5~2mol/L的盐酸、硫酸或高氯酸浸泡6~10h,用乙醇清洗,干燥后得到单斜相铱酸锶SrIrO
或,
(1)混合溶液的配制:称取n
(2)干燥和煅烧:将步骤(1)得到的混合溶液在120~200℃环境中干燥5~10h,将干燥后的固态样品研磨成粉末以0.5~5℃/min的升温速率升温,然后在120~220℃、230~320℃、330~520℃和620~720℃下分别加热1~10h、1~10h、2~4h和1~10h,自然冷却至室温得到黑色粉末;
(3)酸处理:将步骤(2)最终得到的黑色粉末用0.5~2mol/L的盐酸、硫酸或高氯酸浸泡6~10h,用乙醇清洗,干燥后得到正交相铱酸锶SrIrO
或,
(1)混合溶液的配制:称取n
(2)干燥和煅烧:将步骤(1)得到的混合溶液在120~200℃环境中干燥5~10h,将干燥后的固态样品研磨成粉末以0.5~5℃/min的升温速率升温,然后在120~220℃、230~320℃、330~520℃和620~720℃下分别加热1~10h、1~10h、2~4h和1~10h,自然冷却至室温得到黑色粉末;
(3)酸处理:将步骤(2)最终得到的黑色粉末用0.5~2mol/L的盐酸、硫酸或高氯酸浸泡6~10h,用乙醇清洗,干燥后得到立方相Sr
2.如权利要求1所述的一类铱酸锶催化剂的制备方法,其特征在于:铱源为六氯铱(IV)酸钾、六氯铱(III)酸钾、氯化铱、氯铱酸或其混合物。
3.如权利要求1所述的一类铱酸锶催化剂的制备方法,其特征在于:锶源为硝酸锶、氯化锶、氢氧化锶、碳酸锶或其混合物。
4.如权利要求1所述的一类铱酸锶催化剂的制备方法,其特征在于:有机多元醇为乙二醇或丙三醇。
5.如权利要求1所述的一类铱酸锶催化剂的制备方法,其特征在于:有机多元酸为柠檬酸、酒石酸或草酸。
6.一类铱酸锶催化剂,其特征在于:是由权利要求1~5任何一项所述的方法制备得到。
7.权利要求6所述的铱酸锶催化剂在电催化裂解酸性水产氧方面的应用。
说明书
技术领域
本发明属于无机功能材料领域,具体涉及一系列纯相铱酸锶类催化剂、制备方法及其在高效电催化裂解酸性水产氧方面的应用。
背景技术
随着经济社会的飞速发展,人口增多,能源问题日益凸显。在可持续能源发展领域,例如燃料电池和电催化产氢中,电化学水裂解产氧反应至关重要,但是产氧反应动力学限制了其催化性能和商业化应用。在酸性水氧化中,问题更加凸显,大部分催化材料在强酸和产氧过电势下很难稳定。传统的贵金属氧化物(IrO2,RuO2)催化剂具有较高的酸性水氧化(OER)活性,RuO2在电化学酸性水氧化催化中性质优异,但是稳定性很差。IrO2在电化学水氧化中表现良好,但价格昂贵且资源匮乏限制了它的大规模应用。所以寻找取代贵金属铱的氧化物或降低贵金属铱的用量,并具有更好的酸性水氧化产氧活性和稳定性的催化材料是酸性水裂解产氧的关键。
近几年里,随着相关研究的推进,一系列酸性产氧催化剂被开发出来。例如,具有高本征酸性水裂解产氧活性的IrOx/SrIrO3(Science 2016年353卷1011页);铱基双钙钛矿型高效水氧化催化剂(Nat.Commun.2016年7卷12363页)和水热法合成烧绿石结构的Bi2Ir2O7产氧催化剂(Chem.Mater.2014年24卷4192页)。尽管这些催化剂在酸性产氧方面性质较良好,但是其合成方法都较为复杂,大多需要模板诱导晶相合成法或者高温高压固相合成法。针对上述问题,我们用较普适的溶胶凝胶法,常压(101.325kPa)下即可生成,生成物的酸性水氧化活性更为优异且稳定,可作为十分优异的酸性水裂解水氧化催化剂材料。
发明内容
本发明以合成高效酸性水氧化催化剂为目的,合成了一系列不同相的铱酸锶类催化剂,其中单斜相铱酸锶SrIrO3具有最高的酸性产氧活性,形貌尺寸呈纳微米级的六方片,实现了高效酸性水氧化方面的应用;正交相铱酸锶SrIrO3酸性水氧化活性已被报道,在已报道的酸性水氧化催化剂中本征活性最高,传统合成方法较为困难,需要高温高压或者晶相诱导。本发明中我们实现用一种常压(101.325kPa)方法即溶胶凝胶法合成正交相铱酸锶;其中立方相Sr2Ir3O8在已报道的文献中合成方法多为固相合成,且不能合成纯相。本发明通过调控化合物的计量,发现一系列可以合成出纯相Sr2Ir3O8的配比,并且成功合成出纯相纳微米级的立方相Sr2Ir3O8立方块。
本发明的所提到的铱酸锶类催化剂是将铱源、有机多元醇、锶源、有机多元酸和水通过一定比例混合加热蒸干,然后将蒸干的反应物煅烧一段时间,再将煅烧得到的产物用0.5~2mol/L的盐酸、硫酸、高氯酸等浸泡3~10h,即可得到相应化合物。
为得到不同相的铱酸锶,我们固定铱源的摩尔量为na,通过调节锶源的摩尔量nb和有机多元酸的摩尔量nc,得到一系列不同相的铱酸锶化合物;具体操作步骤如下:
一、单斜相铱酸锶SrIrO3催化剂的制备:
(1)混合溶液的配置:称取nc摩尔的有机多元酸、nb摩尔的锶源和na摩尔的铱源,其中,nc:nb:na=8:4:1、8:8:1、8:16:1或8:24:1,然后加入到有机多元醇和水的混合溶液中混合均匀;
(2)干燥和煅烧:将步骤(1)得到的混合溶液在120~200℃环境中干燥5~10h,将干燥后的固态样品研磨成粉末后以0.5~5℃/min的升温速率升温,然后在120~220℃、230~320℃、330~520℃和620~720℃下分别加热1~10h、1~10h、2~4h和1~10h,自然冷却至室温得到黑色粉末;
(3)酸处理:将步骤(2)最终得到的黑色粉末用0.5~2mol/L的盐酸、硫酸、高氯酸等浸泡6~10h,用乙醇清洗,干燥后得到单斜相铱酸锶SrIrO3催化剂。
二、正交相铱酸锶SrIrO3催化剂的制备:
(1)混合溶液的配置:称取nc摩尔的有机多元酸、nb摩尔的锶源和na摩尔的铱源,其中,nc:nb:na=12~40:8:1,然后加入到有机多元醇和水的混合溶液中混合均匀;
(2)干燥和煅烧:将步骤(1)得到的混合溶液在120~200℃环境中干燥5~10h,将干燥后的固态样品研磨成粉末以0.5~5℃/min的升温速率升温,然后在120~220℃、230~320℃、330~520℃和620~720℃下分别加热1~10h、1~10h、2~4h和1~10h,自然冷却至室温得到黑色粉末;
(3)酸处理:将步骤(2)最终得到的黑色粉末用0.5~2mol/L的盐酸、硫酸、高氯酸等浸泡6~10h,用乙醇清洗,干燥后得到正交相铱酸锶SrIrO3催化剂。
三、立方相Sr2Ir3O8立方块催化剂的制备:
(1)混合溶液的配置:称取nc摩尔的有机多元酸、nb摩尔的锶源和na摩尔的铱源,其中,nc:nb:na=0~4:4~10:1,然后加入到有机多元醇和水的混合溶液中混合均匀;
(2)干燥和煅烧:将步骤(1)得到的混合溶液在120~200℃环境中干燥5~10h,将干燥后的固态样品研磨成粉末以0.5~5℃/min的升温速率升温,然后在120~220℃、230~320℃、330~520℃和620~720℃下分别加热1~10h、1~10h、2~4h和1~10h,自然冷却至室温得到黑色粉末;
(3)酸处理:将步骤(2)最终得到的黑色粉末用0.5~2mol/L的盐酸、硫酸、高氯酸等浸泡6~10h,用乙醇清洗,干燥后得到立方相Sr2Ir3O8立方块催化剂。
上述方法中,铱源包括但不限于六氯铱(IV)酸钾、六氯铱(III)酸钾、氯化铱、氯铱酸等或其混合物。
上述方法中,锶源包括但不限于硝酸锶、氯化锶、氢氧化锶、碳酸锶等或其混合物。
上述方法中,有机多元醇溶剂包括但不限于乙二醇、丙三醇等多元醇溶剂。
上述方法中,有机多元酸包括但不限于柠檬酸、酒石酸、草酸等多元羧酸化合物。
上述方法中,nc:nb:na的摩尔比不限于已列出的比例,适当调节三者关系,均可以合成出三种物质的纯相。
有益效果
1.合成工艺简单,实验程序方便可控,制备周期短,重复性好,可大量生产。
2.所得的单斜相铱酸锶SrIrO3结晶度高,呈规则的六边形片,由于晶体结构内部特有的铱氧八面体共面连接结构,优化了铱氧键本身键氧能力太强的缺点,使材料本身具有极好的酸性水裂解水氧化活性和稳定性。可代替传统的酸性水氧化催化剂二氧化铱,在一定程度上降低了贵金属铱的用量,具有广阔的应用前景。
3.所得的正交相铱酸锶SrIrO3为纳微米级的粉体材料,在已有的报道中,该方法合成工艺最为简单可控,样品纯度高。在酸性水裂解产氧中会转变为IrOx/SrIrO3,具有较高的酸性水裂解水氧化活性。
4.所得的立方相Sr2Ir3O8立方块粉体材料具有极高的结晶度和纯度且形貌尺寸均一可调,通过改变铱源的种类,可以实现Sr2Ir3O8立方块尺寸从微米级到纳米级间的转变。在已有的报道中,该发明是首次合成纯相,且方法简单可控。
附图说明
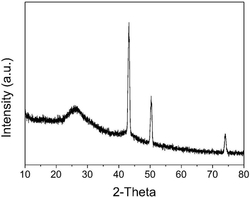
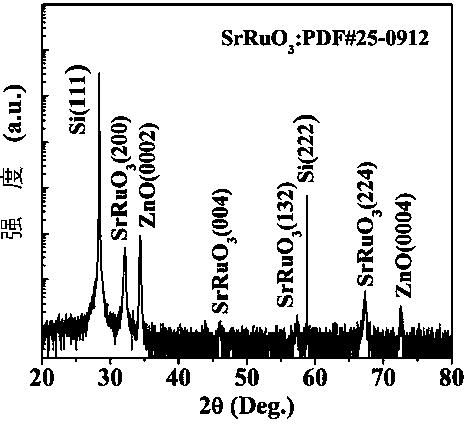
图1:实施例1中单斜相铱酸锶SrIrO3的X射线衍射(XRD)图谱(图A);正交相铱酸锶SrIrO3的X射线衍射(XRD)图谱(图B);立方相Sr2Ir3O8的X射线衍射(XRD)图谱(图C)。
图2:实施例1中单斜相六方片铱酸锶SrIrO3扫描电镜(SEM)照片(图A);正交相块状铱酸锶SrIrO3扫描电镜(SEM)照片(图B);立方块Sr2Ir3O8扫描电镜(SEM)照片(图C)。
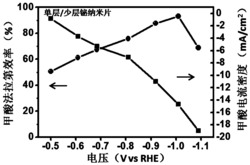
图3:以本发明实施例1产品为水裂解催化剂,在酸性缓冲溶液(0.5M H2SO4)中水裂解析氧(OER)的极化曲线;单斜相铱酸锶SrIrO3水裂解析氧极化曲线(图A);正交相铱酸锶SrIrO3水裂解析氧极化曲线(图B);立方相Sr2Ir3O8水裂解析氧极化曲线(图C)。
图4:以本发明实施例1产品为水裂解催化剂,在酸性缓冲溶液(0.5M H2SO4)中水裂解析氧(OER)的稳定性曲线。单斜相铱酸锶SrIrO3水裂解析氧稳定性曲线(图A);正交相铱酸锶SrIrO3水裂解析氧稳定性曲线(图B);立方相Sr2Ir3O8水裂解析氧稳定性曲线(图C)。
具体实施方式
下面通过实施例并结合附图对本发明作进一步说明,但本发明的保护范围不限于下述的实施例。本领域技术人员清楚,在不偏离本发明主旨和范围的情况下可以对本发明做出变化或调整,这些变化或调整也纳入本发明的保护范围内。
实施例1
单斜相铱酸锶SrIrO3的制备:首先,将80mg(0.166mmol)六氯铱(IV)酸钾放入4mL乙二醇中,室温搅拌至溶解完全,为深棕色透明溶液,称为溶液a。将280mg(1.332mmol)柠檬酸和280mg(1.323mmol)将硝酸锶溶解于5mL蒸馏水中,室温搅拌至无色透明溶液,称为溶液b。将溶液b逐滴滴加到溶液a中,充分搅拌均匀后将混合溶液在180℃环境下蒸干6h。本实施例中,nc:nb:na=8:8:1。然后将蒸干的固态样品研磨成粉末,以1.7℃/min的升温速率在200℃,300℃,500℃和700℃下分别加热6h,6h,3h和6h。随炉冷却后,将黑色粉末用1mol/L的盐酸浸泡6h,用乙醇清洗,干燥后收集样品粉末。
正交相铱酸锶SrIrO3的制备:首先,将80mg(0.166mmol)六氯铱(IV)酸钾放入4mL乙二醇中,室温搅拌至溶解完全,为深棕色透明溶液,称为溶液a。将840mg(3.997mmol)柠檬酸和280mg(1.323mmol)硝酸锶溶解于5mL蒸馏水中,室温搅拌至无色透明溶液,称为溶液b。将溶液b逐滴滴加到溶液a中,充分搅拌均匀后将混合溶液放在180℃环境下蒸干6h。本实施例中,nc:nb:na=24:8:1。然后将蒸干的固态样品研磨成粉末,以1.7℃/min的升温速率在200℃,300℃,500℃和700℃下分别加热6h,6h,3h和6h。随炉等却后,将黑色粉末用1mol/L的盐酸浸泡6h,用乙醇清洗,干燥后收集样品粉末。
立方相Sr2Ir3O8立方块的制备:首先,将80mg(0.166mmol)六氯铱(IV)酸钾放入4mL乙二醇中,室温搅拌至溶解完全,为深棕色透明溶液,称为溶液a。将140mg(0.662mmol)硝酸锶溶解于5mL蒸馏水中,室温搅拌至无色透明溶液,称为溶液b。将溶液b逐滴滴加到溶液a中,充分搅拌均匀后将混合溶液放在180℃环境下蒸干6h。本实施例中,nc:nb:na=0:4:1然后将蒸干的固态样品研磨成粉末,以1.7℃/min的升温速率在200℃,300℃,500℃和700℃下分别加热6h,6h,3h和6h。随炉等却后,将黑色粉末用1mol/L的盐酸浸泡6h,用乙醇清洗,干燥后收集样品粉末。
对上述方法制备的材料在标准三电极电解池中进行电催化水裂解析氧(OER)性质测试;将本发明产品混合在体积含量为10%~50%的萘酚异丙醇溶液中,将溶液滴在铂碳电极上作为电解池中工作电极;参比电极为饱和甘汞电极,对电极为铂丝,电解液为0.5MH2SO4。需要说明的是,电催化测试中所有以饱和甘汞为参比电极得到的电势在性质图中均转换为可逆氢电极电势,外接电源为电化学工作站主电池。
对上述方法制备的材料进行了一些结构和性能研究。图1A为获得的单斜相铱酸锶SrIrO3X射线衍射(XRD)图谱的;图1B为获得的正交相铱酸锶SrIrO3X射线衍射(XRD);图1C为获得的立方相Sr2Ir3O8立方块X射线衍射(XRD)。从图1中我们可以看出,用这种方法获得的铱酸锶类化合物均为纯相。
图2为获得的铱酸锶类化合物的扫描电镜(SEM)照片,图2A表明获得的单斜相铱酸锶直径约为1μm、厚度为约50nm的六边形。图2B表明获得的正交相铱酸锶为微米级的块状样品。图2C表明获得的立方相Sr2Ir3O8正方体边长约为200nm左右的立方块。
图3为本发明产品为水裂解催化剂在酸性硫酸(0.5M H2SO4)溶液中水裂解析氧(OER)的极化曲线。图3A为单斜相铱酸锶SrIrO3水裂解析氧反应极化曲线,在过电势为248mV,达到电流密度为10mA/cm
图4为本发明产品为酸性水裂解催化剂电解液硫酸(0.5M H2SO4)溶液中水裂解析氧(OER)的稳定性曲线。图4A为单斜相铱酸锶SrIrO3水裂解析氧时电流密度保持在10mA/cm
实施例2
与实施例1相同,只是单斜相铱酸锶SrIrO3的制备中将280mg(1.323mmol)硝酸锶减少至140mg(0.662mmol),此时nc:nb:na=8:4:1,其他反应物的量和条件不变。
钙钛矿相铱酸锶SrIrO3的制备中将840mg(3.997mmol)柠檬酸的量减少至420mg(1.999mmol),此时nc:nb:na=12:8:1,其他反应物的量和条件不变。
立方相Sr2Ir3O8的制备中将140mg(0.662mmol)硝酸锶的量增加至350mg(1.654mmol),此时nc:nb:na=0:10:1,其他反应物的量和条件不变。
单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例3
与实施例1相同,只是单斜相铱酸锶SrIrO3的制备中将280mg(1.332mmol)硝酸锶增加为560mg(2.646mmol),此时nc:nb:na=8:16:1,其他反应物的量和条件不变。
钙钛矿相铱酸锶SrIrO3的制备中将840mg(3.969mmol)柠檬酸的量增加至1120mg(5.330mmol),此时nc:nb:na=32:8:1,其他反应物的量和条件不变。
立方相Sr2Ir3O8的制备中加入140mg(0.666mmol)柠檬酸,此时nc:nb:na=4:4:1,其他反应物的量和条件不变。
单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例4
与实施例1相同,只是单斜相铱酸锶SrIrO3的制备中将280mg(1.323mmol)硝酸锶增加为560mg(2.646mmol),此时nc:nb:na=8:16:1,其他反应物的量和条件不变。
钙钛矿相铱酸锶SrIrO3的制备中将840mg(3.997mmol)柠檬酸的量增加至1120mg(5.330mmol),此时nc:nb:na=32:8:1,其他反应物的量和条件不变。
立方相Sr2Ir3O8的制备中加入140mg(0.666mmol)柠檬酸,此时nc:nb:na=4:4:1,其他反应物的量和条件不变。
单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例5
与实施例1相同,只是单斜相铱酸锶SrIrO3的制备中将280mg(1.323mmol)硝酸锶增加为840mg(3.969mmol),此时nc:nb:na=8:24:1,其他反应物的量和条件不变。
钙钛矿相铱酸锶SrIrO3的制备中将840mg(3.997mmol)柠檬酸的量增加至1400mg(6.662mmol),此时nc:nb:na=40:8:1,其他反应物的量和条件不变。
立方相Sr2Ir3O8的制备中加入140mg(0.666mmol)柠檬酸,140mg(0.662mmol)硝酸锶的量提高到350mg(1.654mmol)此时nc:nb:na=4:10:1,其他反应物的量和条件不变。
单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例6
与实施例1相同,只是将六氯铱(IV)酸钾换为六氯铱(III)酸钾,铱源的摩尔数不变,为0.166mmol。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例7
与实施例1相同,只是将六氯铱(IV)酸钾换为氯铱酸水合物,铱源的摩尔数不变,为0.166mmol。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例8
与实施例1相同,只是将不同相的制备中六氯铱(IV)酸钾换为氯化铱,铱源的摩尔数不变,为0.166mmol。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例9
与实施例1相同,只是将不同相的制备中硝酸锶换为氯化锶,锶源的摩尔量不变。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例10
与实施例1相同,只是将不同相的制备中硝酸锶换为氢氧化锶,锶源的摩尔量不变。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例11
与实施例1相同,只是将不同相的制备中硝酸锶换为碳酸锶,锶源的摩尔量不变。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例12
与实施例1相同,只是将不同相的制备中柠檬酸换为酒石酸,制备单斜相铱酸锶SrIrO3酒石酸的摩尔量为1.332mmol。制备正交相铱酸锶SrIrO3酒石酸摩尔量为3.9996mmol。制备立方相Sr2Ir3O8立方块酒石酸摩尔量为0.666mmol。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为260mV时,该材料电流密度达到10mA/cm
实施例13
与实施例1相同,只是将乙二醇换为丙三醇,固定醇的体积为4mL。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为250mV时,该材料电流密度达到10mA/cm
实施例14
与实施例1相同,只是将煅烧温度变为以2℃/min的升温速率在200℃,300℃,500℃和800℃下分别加热6h,6h,3h和6h。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为265mV时,该材料电流密度达到10mA/cm
实施例15
与实施例1相同,只是将煅烧温度变为以1.7℃/min的升温速率在300℃,500℃,600℃和700℃下分别加热6h,6h,3h和6h。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为245mV时,该材料电流密度达到10mA/cm
实施例16
与实施例1相同,只是将煅烧温度变为以1.5℃/min的升温速率在500℃,600℃和700℃下分别加热6h。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为245mV时,该材料电流密度达到10mA/cm
实施例17
与实施例1相同,只是将煅烧温度变为以1℃/min的升温速率在600℃和700℃下分别加热6h。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为245mV时,该材料电流密度达到10mA/cm
实施例18
与实施例1相同,只是将煅烧温度变为以1.7℃/min的升温速率从室温升高至700℃下加热6h。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例19
与实施例1相同,只是将酸处理的1mol/L的盐酸换为0.5M H2SO4溶液。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为245mV时,该材料电流密度达到10mA/cm
实施例20
与实施例1相同,只是将酸处理的1mol/L的盐酸换为1M HClO4溶液。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为245mV时,该材料电流密度达到10mA/cm
实施例21
与实施例1相同,只是将酸性催化剂电解液硫酸(0.5M H2SO4)溶液换为0.1M HClO4溶液。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为255mV时,该材料电流密度达到10mA/cm
实施例22
与实施例1相同,只是将酸性催化剂硫酸(0.5M H2SO4)溶液换为1M H2SO4溶液。所得样品的电催化性能:单斜相铱酸锶SrIrO3,当过电势为240mV时,该材料电流密度达到10mA/cm
铱酸锶类催化剂、制备方法及其在电催化裂解酸性水产氧方面的应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0