

IPC分类号 : B01J23/745,B01J23/80,C06D5/00,C06B29/22








专利摘要
本发明提出一种金属离子掺杂Fe2O3催化材料的制备方法,包括步骤:(1)将三价铁盐溶解于乙醇和/或H2O的溶剂中,不断搅拌,至分散均匀;(2)将金属盐加入步骤(1)所得的分散液中继续搅拌,至分散均匀;(3)将CH3COONa加入分散液中继续搅拌,至分散均匀;(4)将混合溶液转移到内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中进行反应;(5)自然冷却到室温,用水和乙醇清洗。本发明还提出所述的Fe2O3催化材料在催化高氯酸铵热分解反应中的应用。本发明提出的金属离子掺杂Fe2O3纳米材料的制备过程工艺简单、重复性好,掺杂原料Cu2+、Zn2+等廉价易得,有较高的应用前景和使用价值。
权利要求
1.一种金属离子掺杂Fe2O3催化材料的制备方法,其特征在于,包括步骤:
(1)将三价铁盐溶解于乙醇和/或H2O的溶剂中,不断搅拌,至分散均匀;
(2)将金属盐加入步骤(1)所得的分散液中继续搅拌,至分散均匀;所述金属盐为CuCl2、ZnCl2、CuSO4、ZnSO4、CoSO4、NiSO4中的一种或两种;
(3)将CH3COONa加入步骤(2)所得的分散液中继续搅拌,至分散均匀;
(4)将步骤(3)制备的混合溶液转移到内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中进行反应;
(5)将步骤(4)中的反应产物自然冷却到室温,用水和乙醇清洗,放入烘箱中干燥,即可得金属离子掺杂Fe2O3纳米材料。
2.根据权利要求1所述的制备方法,其特征在于,所述步骤(1)中三价铁盐为FeCl3或Fe2(SO4)3,三价铁盐在溶剂中的浓度为0.05~0.1mol/L,所述溶剂为无水乙醇和H2O体积比为10:0~5的溶剂。
3.根据权利要求1所述的制备方法,其特征在于,所述步骤(2)加入的金属盐占三价铁盐的摩尔比例为0.02~0.05。
4.根据权利要求1~3任一项所述的制备方法,其特征在于,所述步骤(3)加入的CH3COONa与三价铁盐的摩尔比例为5~25。
5.根据权利要求1~3任一项所述的制备方法,其特征在于,所述步骤(1)至步骤(3)中,搅拌的时间互相独立地为20min~1h。
6.根据权利要求1~3任一项所述的制备方法,其特征在于,所述步骤(4)水热反应釜的加料系数为40~60%,反应温度为150~180℃,反应时间为4~24h。
7.根据权利要求1~3任一项所述的制备方法,其特征在于,所述步骤(5)中用水和乙醇分别清洗,共清洗4~6次;干燥的温度为50~70℃,干燥时间为5~12h。
8.权利要求1~6任一项所述制备方法制备得到的金属离子掺杂Fe2O3催化材料。
9.权利要求8所述的Fe2O3催化材料在催化高氯酸铵热分解反应中的应用。
说明书
技术领域
本发明属于催化材料领域,具体涉及一种金属掺杂的催化材料的制备方法。
背景技术
高氯酸铵(NH4ClO,简称AP)在常温下为白色斜方晶体,当受热温度超过150摄氏度时,开始分解过程。AP的热分解分三个阶段,350℃以下的低温分解和350~450℃的升华和高温分解。高氯酸铵是复合固体火箭推进剂的重要氧化剂和高能组分,其燃烧和热分解性能对固体火箭推进剂的燃烧过程具有重要影响。对AP热分解性能的改善包括降低它的分解温度,尤其是降低它的高温分解温度,同时增加分解热量,提高热分解速率,提高热分解反应的激烈程度。
通过添加少量催化剂改善AP的热分解性能是行之有效的方法之一。研究表明,纳米金属、纳米过渡金属氧化物等许多纳米材料对AP的热分解具有明显的催化作用,例如氧化铁(III)、一氧化铅(II)纳米材料等,可以使AP高温分解温度下降40~80℃不等。赤铁矿(α-Fe2O3),是氧化铁(III)的主要矿物形式,在自然界分布极广。α-Fe2O3具有成本低廉、抗腐蚀性优良和污染性小等优点,在无机催化剂方面具有广泛的应用前景。近年来,关于不同形貌α-Fe2O3合成与催化性能、α-Fe2O3与碳材料复合材料制备与催化等研究非常活跃。除此之外,掺杂也是提高材料理化性能的方法之一。与金属离子掺杂可以降低材料禁带宽度、提高氧化铁(III)材料的导电性和电化学性能(马荣伟等,《纳米技术》,2014,4(2):22-29;Lu,Guixia,et al.Electrochimica Acta,2014,117:230-238)。具体作为高氯酸铵燃烧反应的促进材料,氧化铁(III)也有望通过掺杂金属离子进一步提高其催化性能。
发明内容
针对本领域研究现状,本发明的目的在于提供一种基于金属离子掺杂纳米Fe2O3的催化材料及其制备方法,为高氯酸铵的热分解提供一种新型高效的催化剂。
本发明的另一目的是提出所述催化剂的应用。
实现本发明上述目的的技术方案为:
一种金属离子掺杂Fe2O3催化材料的制备方法,包括步骤:
(1)将三价铁盐溶解于乙醇和/或H2O的溶剂中,不断搅拌,至分散均匀;
(2)将金属盐加入步骤(1)所得的分散液中继续搅拌,至分散均匀;所述金属盐为CuCl2、ZnCl2、CuSO4、ZnSO4、CoSO4、NiSO4中的一种或两种;
(3)将CH3COONa加入步骤(2)所得的分散液中继续搅拌,至分散均匀;
(4)将步骤(3)制备的混合溶液转移到内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中进行反应;
(5)将步骤(4)中的反应产物自然冷却到室温,用水和乙醇清洗,放入烘箱中干燥,即可得金属离子掺杂Fe2O3纳米材料。
进一步地,所述步骤(1)中三价铁盐为FeCl3或Fe2(SO4)3,三价铁盐在溶剂中的浓度为0.05~0.1mol/L,所述溶剂为无水乙醇和H2O体积比为10:0~5的溶剂。
优选地,所述步骤(2)加入的金属盐占三价铁盐的摩尔比例为0.02~0.05。
其中,所述步骤(3)加入的CH3COONa与三价铁盐的摩尔比例为5~25。
其中,所述步骤(1)至步骤(3)中,搅拌的时间互相独立地为20min~1h。
其中,所述步骤(4)水热反应釜的加料系数为40~60%,反应温度为150~180℃,反应时间为4~24h。
其中,所述步骤(5)中用水和乙醇分别清洗,共清洗4~6次;干燥的温度为50~70℃,干燥时间为5~12h。
本发明所述制备方法制备得到的金属离子掺杂Fe2O3催化材料。
所述的Fe2O3催化材料在催化高氯酸铵热分解反应中的应用。
本发明与现有技术相比,具有以下有益效果:
本发明提出的金属离子掺杂Fe2O3纳米材料的制备过程工艺简单、重复性好,掺杂原料Cu2+、Zn2+等廉价易得,有较高的应用前景和使用价值。
本发明制备的金属离子掺杂Fe2O3纳米材料,可用于催化高氯酸铵热分解反应,为高氯酸铵的热分解反应提供了一种新型的催化剂。
本发明制备的Fe2O3纳米材料,其中Cu2+或Zn2+掺杂能够更显著地降低AP的高温热分解温度,利用金属Cu2+、Zn2+通过水热法掺杂α-Fe2O3,改善α-Fe2O3的理化性质,提高α-Fe2O3催化高氯酸铵热分解的性能,与纯AP的高温热分解温度相比可降低115℃。
附图说明
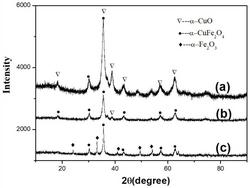
图1为实施例1和6制备的掺杂Fe2O3纳米材料的XRD结果,
图2为实施例1和6制备的掺杂Fe2O3纳米材料的Raman结果,
图3为实施例1制备的掺杂Fe2O3纳米材料的SEM结果,
图4为实施例1制备的掺杂Fe2O3纳米材料的TEM结果,
图5为实施例1制备的掺杂Fe2O3纳米材料的EDS结果,
图6为实施例6制备的掺杂Fe2O3纳米材料的SEM结果,
图7为实施例6制备的掺杂Fe2O3纳米材料的TEM结果,
图8为实施例6制备的掺杂Fe2O3纳米材料的EDS结果,
图9为实施例1和6制备的掺杂Fe2O3纳米材料催化高氯酸铵热分解结果。
图10为实施例2制备的掺杂Fe2O3纳米材料的SEM结果,
图11为实施例3制备的掺杂Fe2O3纳米材料的SEM结果,
图12为实施例4制备的掺杂Fe2O3纳米材料的SEM结果,
图13为实施例5制备的掺杂Fe2O3纳米材料的SEM结果。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。
实施例中,如无特别说明,所采用的手段均为本领域公知的技术手段。
实施例1:
(1)将2mmol FeCl3·6H2O溶解于20ml无水乙醇(分析纯)和5ml H2O的混合溶剂中,磁力搅拌30min,至分散均匀。原料配比见表1。
(2)将0.05mmol CuCl2加入步骤(1)所得的分散液中,继续搅拌30min,至分散均匀。
(3)将20mmol CH3COONa加入步骤(2)所得的分散液中,继续搅拌30min,至分散均匀。
(4)将步骤(3)制备的混合溶液转移到体积为50mL、内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中180℃反应12h。
(5)将步骤(4)中的反应产物自然冷却到室温,用水和乙醇分别清洗3次(共清洗6次),放入烘箱中60℃干燥6h,即可得Cu2+掺杂Fe2O3纳米材料。
将实施例1制备的Cu2+掺杂Fe2O3纳米材料经X-射线扫描仪扫描,见图1,衍射峰对应α-Fe2O3的特征衍射峰。将实施例1制备的Cu2+掺杂Fe2O3纳米材料进行拉曼光谱(Raman)分析,见图2拉曼峰对应Fe2O3的特征峰。将实施例1制备的Cu2+掺杂Fe2O3纳米材料通过扫描电子显微镜观察,见图3。将实施例1制备的Cu2+掺杂Fe2O3纳米材料通过透射电子显微镜观察,见图4。显微观察证实本方法制得的材料尺寸在50~100nm范围内。将实施例制备的Cu2+掺杂Fe2O3纳米材料通过X射线能谱仪(EDS)进行元素分析,见图5,表明复合材料含有C、O、Fe、Cu元素。
将实施例1制备的Cu2+掺杂Fe2O3纳米材料用于高氯酸铵热分解。取实施例1制备的0.02g Cu2+掺杂Fe2O3纳米材料和0.48g高氯酸铵在玛瑙研磨中研磨15min至混合均匀,进行热重分析(TG)和示差扫描量热分析(DSC)。测试条件:氩气氛围,升温速率10℃/min,温度范围50~500摄氏度。测试结果见图9,曲线表明,加入4%的Cu2+掺杂Fe2O3纳米材料作为催化剂后,AP的高温分解温度为342℃,比纯AP的高温热分解温度降低115℃,比以Fe2O3纳米材料为催化剂的AP的高温热分解温度降低46℃。
对比例
以FeCl3·6H2O为原料,水热法制备α-Fe2O3纳米材料。
(1)将2mmol FeCl3·6H2O溶解于20ml乙醇和5ml H2O的混合溶剂中,磁力搅拌30min,至分散均匀。
(2)将20mmol CH3COONa加入步骤(1)所得的分散液中,继续搅拌30min,至分散均匀。
(3)将步骤(2)制备的混合溶液转移到体积为50mL、内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中180℃反应12h。
其他操作同实施例1。产物的XRD衍射图谱见图1,衍射峰对应α-Fe2O3的特征衍射峰。拉曼光谱(Raman)分析见图2。
按照和实施例1同样的方法进行热分析试验,差热分析曲线(图9)表明,加入4%的α-Fe2O3纳米材料作为催化剂后,AP的高温分解温度为388℃。
实施例2
(1)将2mmol FeCl3·6H2O溶解于20ml无水乙醇(分析纯)中,磁力搅拌30min,至分散均匀。
其他操作同实施例1。
表1实施例1和实施例2的物料配比
SEM和TEM观察材料的尺寸和形貌和实施例1稍有不同。实施例2SEM图如图10,和实施例1的形貌比较,形状不规则,尺寸也较大。
实施例3
按照和实施例1相同的原料,步骤(4)为:将步骤(3)制备的混合溶液转移到体积为50mL、内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中180℃反应6h。
其他操作同实施例1。
实施例4
按照和实施例1相同的原料,步骤(4)为:将步骤(3)制备的混合溶液转移到体积为50mL、内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中180℃反应9h。
其他操作同实施例1。
实施例5
按照和实施例1相同的原料,步骤(4)为:将步骤(3)制备的混合溶液转移到体积为50mL、内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中180℃反应15h。
其他操作同实施例1。
实施例3-5得到的材料SEM观察见图11-13。和水热反应时间12小时的材料相比,形貌表征不太理想,其预期性能较差。实施例1、3、4、5得到的材料样品的XRD结果没有明显区别。
实施例6:
(1)将2mmol FeCl3·6H2O溶解于20ml乙醇和5ml H2O的混合溶剂中,磁力搅拌30min,至分散均匀。
(2)将0.05mmol ZnCl2加入步骤(1)所得的分散液中,继续搅拌30min,至分散均匀。
(3)将20mmol CH3COONa加入步骤(2)所得的分散液中,继续搅拌30min,至分散均匀。
(4)将步骤(3)制备的混合溶液转移到体积为50ml、内衬为聚四氟乙烯的水热反应釜中,封装后放入烘箱中180℃反应12h。
(5)将步骤(4)中的反应产物自然冷却到室温,分别用水和乙醇清洗3次,放入烘箱中60℃干燥6h,即可得Zn2+掺杂Fe2O3纳米材料。
将实施例6制备的Zn2+掺杂Fe2O3纳米材料经X-射线扫描仪扫描,见图1,衍射峰对应Fe2O3的特征衍射峰。将实施例6制备的Zn2+掺杂Fe2O3纳米材料进行拉曼光谱(Raman)分析,见图2拉曼峰对应Fe2O3的特征峰。将实施例6制备的Zn2+掺杂Fe2O3纳米材料通过扫描电子显微镜观察,见图6。将实施例3制备的Zn2+掺杂Fe2O3纳米材料通过透射电子显微镜观察,见图7。将实施例3制备的Zn2+掺杂Fe2O3纳米材料通过X射线能谱仪(EDS)进行元素分析,见图8,表明复合材料含有C、O、Fe、Zn元素。
将实施例6制备的Zn2+掺杂Fe2O3纳米材料用于高氯酸铵热分解。取实施例6制备的0.02g Zn2+掺杂Fe2O3纳米材料和0.48g高氯酸铵在玛瑙研磨中研磨15min至混合均匀,进行热重分析(TG)和示差扫描量热分析(DSC)。测试条件:氩气氛围,升温速率10℃/min,温度范围50~500摄氏度。测试结果见图9,曲线表明,加入4%的Zn2+掺杂Fe2O3纳米材料作为催化剂后,AP的高温分解温度为342℃,比纯AP的高温热分解温度降低107℃,比Fe2O3纳米材料为催化剂的AP的高温热分解温度降低38℃。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变形,这些改进和变形也应视为本发明的保护范围。
金属离子掺杂Fe2O3催化材料的制备及其应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。



















动态评分
0.0