









专利摘要
本发明属于多酸基稀土材料技术领域,提供了一种Eu/Tb/Tm三元稀土掺杂的多酸衍生物,该多酸衍生物分子式为[N(CH3)4]3K2[EuxTbyTm1‒x‒y(C7H5O2)(H2O)2(α‑PW11O39)]·11H2O(0.02≤x≤0.05,0.40≤y≤0.45)。本发明通过将EuCl3·6H2O、TbCl3·6H2O、TmCl3·6H2O、苯甲酸和多酸前驱体K14[P2W19O69(H2O)]·24H2O溶于蒸馏水中,调节pH至4.5‑4.8,室温下搅拌20―30min,然后在60±5℃条件下加热1.0―1.5h,再趁热加入四甲基氯化铵并搅拌反应20―30min,反应结束后冷却、过滤,滤液静置析出无色块状晶体,即为三元稀土掺杂的多酸衍生物。在该稀土衍生物中,多酸钨簇片段和苯甲酸配体都能够敏化稀土离子发光,Eu3+离子具有红光发射性能,Tb3+离子具有绿光发射性能,Tm3+离子具有绿光发射性能,三原色按照一定比例能够调谐出白光发射。
权利要求
1.一种三元稀土掺杂的多酸衍生物作为荧光发光材料的应用,其特征在于,该多酸衍生物分子式为[N(CH
说明书
技术领域
本发明属于多酸基稀土材料技术领域,具体涉及一种三元稀土掺杂的的多酸衍生物及其制备方法和作为荧光发光材料的应用。
背景技术
基于镧系离子的发光材料,作为经典的无机光学材料,由于其具有潜在的高量子效率、高寿命、可靠性和安全性等优点,多年来一直受到科研工作者和工业界的关注。基于以上优点,稀土基发光材料已用于各种领域,如发光二极管的光学应用、检测与传感、激光、生物成像、生物医学等。
众所周知,多金属氧酸盐(简称多酸),是由高价态的前过渡金属(如:V
近些年来,一系列单核、双核、三核、四核到二十四核的Ln
发明内容
本发明的目的在于制备一种Eu
为实现上述目的,本发明采用如下技术方案:
一种三元稀土掺杂的多酸衍生物,该多酸衍生物分子式为[N(CH3)4]3K2[EuxTbyTm1‒x‒y(C7H5O2)(H2O)2(α-PW11O39)]·11H2O,其中,0.02≤x≤0.05,0.40≤y≤0.45。
上述三元稀土掺杂的多酸衍生物的制备方法,其具体包括以下步骤:
将EuCl3·6H2O、TbCl3·6H2O、TmCl3·6H2O、苯甲酸和多酸前驱体K14[P2W19O69(H2O)]·24H2O 溶于蒸馏水中,调节pH至4.5-4.8,室温下搅拌20―30 min,然后在60 ± 5℃条件下加热1.0―1.5h,再趁热加入四甲基氯化铵并搅拌反应20―30 min,反应结束后冷却、过滤,滤液静置析出无色块状晶体,即为目标产物三元稀土掺杂的多酸衍生物。
其中,多酸前驱体K14[P2W19O69(H2O)]·24H2O参照文献(C. M. Tourne, G. F.Tourne.
具体的,原料EuCl3·6H2O、TbCl3·6H2O和TmCl3·6H2O的质量比为0.005-0.010g:0.095-0.100 g:0.123 g;原料(EuCl3·6H2O+TbCl3·6H2O+TmCl3·6H2O,即三者的摩尔数之和)、苯甲酸、K14[P2W19O69(H2O)]·24H2O和四甲基氯化铵的摩尔比为0.600:0.200:0.465-0.500:1.000。
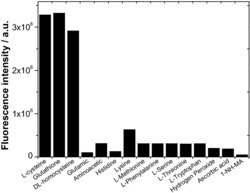
本发明所述Eu
本发明采用自组装合成策略方法,将EuCl3·6H2O/TbCl3·6H2O/TmCl3·6H2O、苯甲酸和多酸前驱体K14[P2W19O69(H2O)]·24H2O按一定摩尔比在水溶液中反应制得具有白光发射性能的三元稀土掺杂多酸衍生物。在该反应中,Eu
1)本发明采用X射线单晶衍射的技术,能够精确解析出多酸稀土衍生物的晶体结构;
2)本发明采用常规水溶液法,操作简单、成本低、安全性高、易于合成且产量较高;通过自组装的合成策略,一步合成目标产物,操作简单、成本较低、环境污染小且安全性高;
3)本发明采用Eu
4)相对于传统的多酸稀土发光材料通过钨簇传递能量到稀土离子,本发明由有机基团起主要敏化稀土离子荧光发射的作用,具有更高的能量传递效率。
附图说明
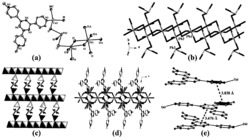
图1是目标产物1‒5的合成示意图;
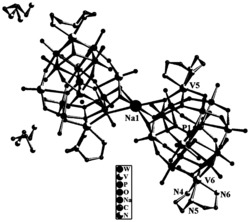
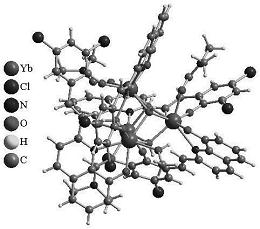
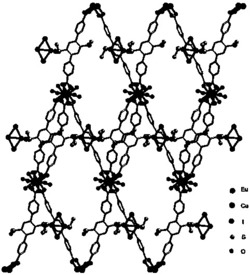
图2是目标产物1‒5阴离子结构的球棍示意图。表明稀土离子Ln
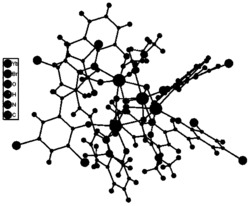
图3是目标产物1‒5阴离子结构的多面体示意图;
图4是目标产物1‒5的热重曲线。目标产物1‒5具有相似的热失重曲线;
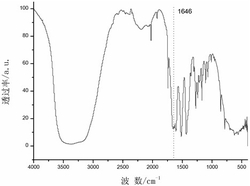
图5是目标产物1‒5的红外光谱图。证明了目标产物1‒5中含有磷钨酸骨架和多酸配体,也证明了目标产物1‒5结构相似;
图6是目标产物1‒5的XRD的模拟图和实验对比图。证明了目标产物1‒5是纯净的,同时证明了目标产物1‒5是同构的;
图7是目标产物1在272 nm激发光下的发射光谱图。证明了目标产物1中Eu荧光发射中心能够在272激发光下被苯甲酸配体激发;
图8是目标产物2在272 nm激发光下的发射光谱图。证明了目标产物2中Tb荧光发射中心能够在272激发光下被苯甲酸配体激发;
图9是目标产物3在272 nm激发光下的发射光谱图。证明了目标产物3中Tm荧光发射中心能够在272激发光下被苯甲酸配体激发;
图10是目标产物4和5在272 nm激发光下的发射光谱图。证明了目标产物4和5中Eu、Tb、Tm荧光发射中心能够在272 nm激发光下被苯甲酸配体激发;
图11是目标产物1‒3的发射光谱对应CIE坐标图。表明目标产物1发射出红色,目标产物2发射出绿色,目标产物3发射出蓝色;
图12是目标产物4和5的发射光谱对应CIE坐标图。表明目标产物4和5发射出白光。
具体实施方式
下面通过实施进一步详细描述本发明,但这并非是对本发明的限制,根据本发明的基本思想,可以做出各种修改和改进,但只要不脱离本发明的基本思想,均在发明的范围之内。
实施例1:
一种含Eu稀土的多酸衍生物的制备方法,其具体包括以下步骤:
将EuCl3∙6H2O (0.228 g,0.600 mmol)、苯甲酸 (0.244 g,0.200 mmol)和多酸前驱体K14[P2W19O69(H2O)]∙24H2O (2.120 g,0.465 mmol)加入到30 mL蒸馏水中,搅拌至完全溶解。用3 mol/L的KOH水溶液将pH值调至4.5,室温下搅拌20―30 min。将该溶液放入60 ℃的水浴中搅拌加热1.5 h,再趁热加入四甲基氯化铵(0.110 g,1.000 mmol)并搅拌反应20―30min,反应结束后待溶液冷却、过滤,滤液静置两周后析出无色块状晶体,即得目标产物1。
实施例2:
一种含Tb稀土的多酸衍生物的制备方法,其具体包括以下步骤:
将TbCl3∙6H2O (0.228 g,0.600 mmol)、苯甲酸 (0.244 g,0.200 mmol)和多酸前驱体K14[P2W19O69(H2O)]∙24H2O (2.120 g,0.465 mmol) 加入到30 mL蒸馏水中,搅拌至完全溶解。用3 mol/L的KOH溶液将pH值调至4.5,室温下搅拌20―30 min。将该溶液放入60 ℃的水浴中搅拌加热1.5 h,再趁热加入四甲基氯化铵(0.110 g,1.000 mmol) 并搅拌反应20―30min,反应结束后待溶液冷却、过滤,滤液静置两周后析出无色块状晶体,即得目标产物2。
实施例3:
一种含Tm稀土的多酸衍生物的制备方法,其具体包括以下步骤:
将TmCl3∙6H2O (0.228 g,0.600 mmol)、苯甲酸(0.244 g,0.200mmol)和多酸前驱体K14[P2W19O69(H2O)]∙24H2O (2.120g,0.465mmol) 加入到30 mL蒸馏水中,搅拌至完全溶解。用3mol/L的KOH溶液将pH值调至4.5,室温下搅拌20―30 min。将该溶液放入60 ℃的水浴中搅拌加热1.5 h,再趁热加入四甲基氯化铵 (0.110 g,1.000 mmol) 并搅拌反应20―30 min,反应结束后待溶液冷却、过滤,滤液静置两周后析出无色块状晶体,即得目标产物3。
实施例4:
一种Eu/Tb/Tm三元稀土掺杂的多酸衍生物的制备方法,其具体包括以下步骤:
将EuCl3∙6H2O(0.010 g,0.026 mmol)、TbCl3∙6H2O (0.095 g,0.254 mmol)和TmCl3∙6H2O(0.123 g,0.320 mmol)的混合物、苯甲酸(0.244g,0.200mmol)、多酸前驱体K14[P2W19O69(H2O)]∙24H2O (2.12 0g,0.465 mmol) 加入到30 mL蒸馏水中,搅拌至完全溶解。用3 mol/L的KOH溶液将pH值调至4.5,室温下搅拌20―30 min。将该溶液放入60 ℃的水浴中搅拌加热1.5 h,再趁热加入四甲基氯化铵(0.110 g,1.000 mmol)并搅拌反应20―30 min,反应结束后待溶液冷却、过滤,滤液静置两周后析出无色块状晶体,即得目标产物4。
实施例5:
一种Eu/Tb/Tm三元稀土掺杂的多酸衍生物的制备方法,其具体包括以下步骤:
将EuCl3∙6H2O(0.005 g,0.013 mmol)、TbCl3∙6H2O(0.100 g,0.267 mmol)和TmCl3∙6H2O(0.123 g,0.320 mmol)的混合物、苯甲酸(0.244g,0.200mmol)、多酸前驱体K14[P2W19O69(H2O)]∙24H2O(2.120g,0.465mmol) 加入到30 mL蒸馏水中,搅拌至完全溶解。用3 mol/L的KOH溶液将pH值调至4.5,室温下搅拌20―30 min。将该溶液放入60℃的水中水浴搅拌加热1.5 h,再趁热加入四甲基氯化铵(0.110 g,1.000 mmol) 并搅拌反应20―30 min,反应结束后待溶液冷却、过滤,滤液静置两周后析出无色块状晶体,即得目标产物5。
实施例6:
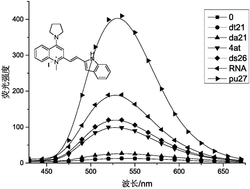
目标产物1的固态荧光光谱在室温下测得。在272 nm的激发光照射下,目标产物1的发射光谱如图11所示,其对应于CIE坐标图上的坐标(0.663,0.335),坐落在红光区。
实施例7:
目标产物2的固态荧光光谱在室温下测得。在272 nm的激发光照射下,目标产物2的发射光谱如图11所示,其对应于CIE坐标图上的坐标(0.324,0.591),坐落在绿光区。
实施例8:
目标产物3的固态荧光光谱在室温下测得。在272 nm的激发光照射下,目标产物3的发射光谱如图11所示,其对应于CIE坐标图上的坐标(0.217,0.210),坐落在蓝光区。
实施例9:
目标产物4的固态荧光光谱在室温下测得。在272 nm的激发光照射下,目标产物4的发射光谱如图12所示,其对应于CIE坐标图上的坐标(0.299,0.337),坐落在白光区。
实施例10:
目标产物5的固态荧光光谱在室温下测得。在272 nm的激发光照射下,目标产物5的发射光谱如图12所示,其对应于CIE坐标图上的坐标(0.279,0.314),坐落在白光区。
本发明通过自组装的方法制备合成了目标产物1‒5(合成路线见图1)。在水溶液中,稀土离子容易水解产生沉淀,加入苯甲酸配体可以起到防止稀土离子水解的作用。多酸前驱体K14[P2W19O69(H2O)]·24H2O在水溶液中能够分解为{PW9O33}和{WO6}多酸片段,有助于构筑结构新颖的多酸构筑块。在该体系中,苯甲酸配体与稀土离子配位后,并与多酸片段反应,形成苯甲酸-稀土离子-多酸多酸稀土衍生物。
本发明采用X-射线单晶衍射技术对上述实施制备得到的目标产物1‒3的晶体结构进行测定和表征,其晶胞参数如下:
目标产物1,单斜晶系,空间群为P-1,晶胞参数a =12.9454 (14) Å, b = 13.5128(14) Å, c = 20.206 (2) Å, α = 82.9783 (18)°, β = 78.1714 (18)°, γ = 75.1355(18)°, V = 3334.7 (6) Å
目标产物2,单斜晶系,空间群为P-1,晶胞参数a = 12.921 (3) Å, b = 13.504 (3)Å, c = 20.156 (4) Å, α = 82.918 (4)°, β = 78.077 (4)°, γ = 75.141 (3)°, V =3316.9 (12) Å
目标产物3,单斜晶系,空间群为P-1,晶胞参数a =12.8403 (14) Å, b = 13.4246(15) Å, c = 19.981 (2) Å, α = 83.917 (2)°, β = 78.657 (2)°, γ = 75.480 (2)°,V = 3263.6 (6) Å
目标产物1‒3具有相似的晶胞参数,表明三者结构是同构的。
同时,本发明对目标产物1‒5进行了红外光谱和XRD表征,目标产物1‒5的红外光谱以及它们XRD图谱的一致性,证实了目标产物1‒5都是同构的。
图4是目标产物1‒5的热重曲线。本发明中,目标产物1‒5具有相似的热失重曲线(图4)。以目标产物1为例,在25‒140°C范围内第一步失重6.17%,对应于11个结晶水的失去(理论值6.05%);在140‒490°C范围内第二步失重7.83%,对应于配位水和有机阳离子氧化分解(理论值7.38%);在490‒890°C范围内第三步失重,对应于有机配体部分氧化分解和多酸骨架的部分分解。
本发明中对目标产物1‒5进行了红外光谱和XRD表征(见图5和6)。在红外光谱图中,目标产物1‒5具有相似的红外光谱谱图,700‒1100 cm
通过红外光谱和XRD表征证明:目标产物1‒5结构是同构的,在此,以目标产物1为例,对目标产物1‒5的结构进行详细描述(详见图2和3)。由图2和3 可以看出:目标产物1包括一个[Eu(C7H5O2)(α-PW11O39)]
本发明中,在272nm的激发光下,
目标产物1结构中的苯甲酸配体和多酸组分吸收能量,然后传递给Eu
目标产物2结构中的苯甲酸配体和多酸组分吸收能量,然后传递给Tb
目标产物3结构中的苯甲酸配体和多酸组分吸收能量,然后传递给Tm
目标产物4结构中的苯甲酸配体和多酸组分吸收能量,然后传递给Eu
目标产物5结构中的苯甲酸配体和多酸组分吸收能量,然后传递给Eu
一种三元稀土掺杂的多酸衍生物及其制备方法和作为荧光发光材料的应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0