








专利摘要
本发明提供了一种氧桥双核烯丙基硅桥氨基锆催化剂及其制备方法和应用,涉及乙烯聚合IVB过渡金属催化剂,具体是一种具有N‑C‑C‑Si‑N骨架的烯丙基配体锆配合物催化剂。其制备方法:在氮气的保护下,用二异丙基胺基锂(LDA)将有取代基的酮亚胺去氢,加入等摩尔的溴化镁得到镁盐,随后加入等摩尔的硅烷化合物得到中性化合物,经过二次锂化制备烯丙基硅桥氨基锂化合物L,最后与ZrCl4反应并通入适量氧气得到氧桥双核烯丙基硅桥氨基锆催化剂C。该制备方法简单,用料简单易得、价格低廉;催化剂用于乙烯聚合显示高的催化活性。
权利要求
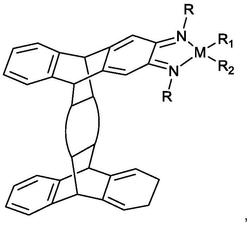
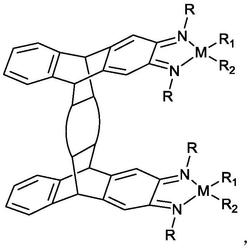
1.一种氧桥双核烯丙基硅桥氨基锆催化剂,其特征在于,结构式为:
其中:R1、R2各自独立地选自自氢和1-10个碳原子的烷基、芳基、烷芳基、芳烷基和烷氧基中的一种;X为卤素,优选氯或溴。
2.如权利要求1所述的一种氧桥双核烯丙基硅桥氨基锆催化剂,其特征在于,所述的R1独立地选自氢,甲基,乙基,异丙基和芳基中的一种;R2独立地选甲基,苯基中的一种。
3.如权利要求1或2所述的一种氧桥双核烯丙基硅桥氨基锆催化剂的制备方法,其特征在于,包括如下步骤:
(1)锂盐的制备:在氮气的保护下,丙酮浴下,将酮亚胺有机化合物和二异丙基胺基锂按照等摩尔反应,溶剂为四氢呋喃,反应1-3小时后,再次冷却至-78℃加入等摩尔的溴化镁,当溶液自然升温到室温以后,保持搅拌反应8-10小时,得到镁盐的四氢呋喃溶液,在氮气的保护下,冰水浴下,将等摩尔的硅烷化合物缓慢加入其中,当溶液自然升温到室温以后,保持搅拌反应10-12小时,用甲苯萃取得到油状的中性化合物;然后在氮气的保护下,冰水浴下,用等摩尔的丁基锂再次锂化,保持搅拌反应8-10小时,在四氢呋喃中结晶得到无色烯丙基硅桥氨基锂L;
(2)催化剂的制备:在氮气的保护下,在丙酮浴下,把ZrCl4加入到烯丙基硅桥氨基锂L的四氢呋喃溶液中,ZrCl4与L的摩尔比为1:1-2,当溶液自然升温到室温以后,注入适量的氧气,保持搅拌反应20-24小时,真空抽干,加入二氯甲烷,过滤,将滤液浓缩得到氧桥双核烯丙基硅桥氨基锆催化剂。
4.如权利要求1或2所述的一种氧桥双核烯丙基硅桥氨基锆催化剂在乙烯聚合中的应用。
说明书
技术领域
本发明涉及乙烯聚合IVB过渡金属催化剂,具体而言涉及一种具有N-C-C-Si-N骨架的烯丙基配体锆配合物催化剂,更具体地说是一种氧桥双核烯丙基硅桥氨基锆催化剂及其制备方法和应用。
背景技术
目前,聚乙烯树脂是通用合成树脂中产量最大的品种,因其价格低、性能好等特点,在我国的应用相当广泛,如薄膜、注塑制品、电线电缆、中空制品等都在其消费结构中占有较大的比例。工业化的烯烃聚合催化剂有Ziegler-Natta型催化剂、Phillips型催化剂和茂金属型等催化剂,这些催化剂可以通过调节配体的结构来达到对催化活性以及聚合物性能的控制。其中氮杂烯丙基过渡金属配合物催化剂的报道非常少,而且存在制备原料工艺复杂,价格昂贵,产率很低,催化活性低等缺点。
发明内容
本发明的目的是提供一种氧桥双核烯丙基硅桥氨基锆催化剂及其制备方法,以及该催化剂在乙烯聚合中的应用。催化剂制备方法简单、原料简单易得、价格低廉,催化剂用于乙烯聚合应有高的催化活性。
本发明提供的一种氧桥双核烯丙基硅桥氨基锆催化剂,具有如下的结构式:
其中:
R1、R2各自独立地选自氢和1-10个碳原子的烷基、芳基、烷芳基、芳烷基和烷氧基中的一种;R1独立地优选自氢,甲基,乙基,异丙基和芳基中的一种;R2独立地优选甲基,苯基中的一种;
X为卤素,优选氯或溴。
本发明还提供了一种氧桥双核烯丙基硅桥氨基锆催化剂的制备方法,包括如下步骤:
(1)锂盐的制备:在氮气的保护下,丙酮浴下,将酮亚胺有机化合物和二异丙基胺基锂按照等摩尔反应,溶剂为四氢呋喃,反应1-3小时后,再次冷却至-78℃加入等摩尔的溴化镁,当溶液自然升温到室温以后,保持搅拌反应8-10小时,得到镁盐的四氢呋喃溶液,在氮气的保护下,冰水浴下,将等摩尔的硅烷化合物缓慢加入其中,当溶液自然升温到室温以后,保持搅拌反应10-12小时,用甲苯萃取得到油状的中性化合物;然后在氮气的保护下,冰水浴下,用等摩尔的丁基锂再次锂化,保持搅拌反应8-10小时,在四氢呋喃中结晶得到无色烯丙基硅桥氨基锂L;
(2)催化剂的制备:在氮气的保护下,在丙酮浴下,把ZrCl4加入到烯丙基硅桥氨基锂L的四氢呋喃溶液中,ZrCl4与L的摩尔比为1:1-2,当溶液自然升温到室温以后,注入适量的氧气,保持搅拌反应20-24小时,真空抽干,加入二氯甲烷,过滤,将滤液浓缩得到氧桥双核烯丙基硅桥氨基锆催化剂C。具体合成路线如下:
氧桥双核烯丙基硅桥氨基锆催化剂可以在乙烯聚合中的应用。在乙烯聚合的实验中催化剂聚合活性最高可达:7.23×105g·mol(Zr)-1·h-1,聚乙烯分子量可达:9.58×105g·mol-1。
与现有技术相比本发明的优点和效果:合成催化剂所用原料简单易得、价格低廉,制备方法简单;催化剂用于乙烯聚合有高的催化活性,可得到具有高分子量的聚乙烯。
附图说明
图1为催化剂C1的晶体结构;
具体实施方式
下面仅仅为说明而给出的实施例,这些实施例并非用于限制本发明的保护范围。
实施例1
(1)锂盐的制备
在氮气的保护下,冰水浴下,将二甲基氨基二甲基硅基氯化物(0.3ml,2mmol)缓慢加入到2,6-二乙基-4-甲基-N-苯乙烯基苯胺基溴化镁的四氢呋喃配合物(2mmol)的四氢呋喃溶液中,当溶液自然升温到室温以后,保持搅拌反应6小时,用甲苯萃取得到油状的N-(2-二甲基氨基二甲基硅基-1-苯乙烯基)-2,6-二乙基-4-甲基苯胺中性化合物。然后在氮气的保护下,丙酮浴下,用二异丙基胺基锂(0.21g,2mmol)再次锂化,保持搅拌反应6小时,在四氢呋喃中结晶得到无色N-(2-二甲基氨基二甲基硅基-1-苯乙烯基)-2,6-二乙基-4-甲基苯胺基锂的四氢呋喃配合物L1;
中间体L1的产率及其表征的数据如下:
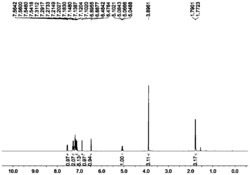
L1:(R1=Me)黄色油状物:产率99%,1H NMR(300MHz CDCl3):δ0.01(d,6H,Si(CH3)2),1.21–1.25(t,6H,CH2CH3),2.32(s,3H,CH3),2.57–2.61(q,4H,CH2CH3),2.44(d,6H,N(CH3)2),4.01(s,H,NH),4.62(s,H,CH),7.24–7.30,7.53–7.57(m,5H,Ph),7.79(s,2H,m-Ar).
L2:(R1=H)淡黄色油状物:产率98%,1H NMR(300MHz CDCl3):δ0.01(d,6H,Si(CH3)2),1.23–1.27(t,6H,CH2CH3),2.58–2.63(q,4H,CH2CH3),2.49(d,6H,N(CH3)2),3.98(s,H,NH),4.52(s,H,CH),7.34–7.40,7.50(m,5H,Ph),7.69,7.91(s,2H,m,p-Ar).
(2)催化剂C的制备:将Schlenk瓶抽真空通N2置换三次以后,加入L(2mmol)和四氢呋喃25ml,在-78℃加入ZrCl4(2mmol)搅拌下恢复室温,注入氧气反应24小时,真空抽干加入二氯甲烷50毫升,过滤,浓缩,正己烷洗涤,二氯甲烷中结晶,得到黄色晶体化合物C1(R1=Me),产率:43%。1H NMR(300MHz CDCl3)δ0.01,0.21(s,12H,Si(CH3)2),1.13,1.25,1.31,1.35(m,12H,CH2CH3),1.79,1.82(m,6H,CH3),2.16(s,12H,N(CH3)2),2.36,2.49(m,8H,CH2CH3),3.89(d,2H,NH),4.19(d,2H,CH),6.95,6.98–7.15(m,10H,Ph),7.24,7.34–7.54(m,4H,Ar).13C NMR(75MHz CDCl3)δ-1.10,-0.65(Si(CH3)2),24.09,28.71,31.23,32.16(CH2CH3),33.14,34.25(CH2CH3),32.19,32.54(CH3),40.17,41.28(N(CH3)2),82.27(CH),123.6–125.4,127.9–133.1(Ar and Ph),135.6(ipso-CPh),140.0,143.5(ipso-CAr),150.8(NCC).晶体结构见图1。
亮黄色晶体化合物C2(R1=H),产率49%。1H NMR(300MHz CDCl3)δ0.03,0.31(q,6H,Si(CH3)2),0.58–0.63,1.29–1.34(t,6H,CH2CH3),2.46(d,6H,N(CH3)2),3.65–3.79(q,4H,CH2),4.01(s,H,NH),4.14(s,H,CH),6.74–6.89(m,5H,Ph),7.34–7.39(m,3H,Aryl).13C NMR(75MHz CDCl3)δ-1.36,-0.48(Si(CH3)2),22.74,24.01(CH2CH3),34.14(CH2CH3),40.13,42.85(N(CH3)2),81.68(CH),118.1,121.8,123.5,125.2,127.8(Aryl and Ph),134.2(ipso-CPh),138.6,145.9(ipso-CAryl),153.8(NCC).
实施例2
1.催化剂C1的制备同实施例1。
2.乙烯聚合:将装有机械搅拌和热电耦的250毫升的不锈钢釜抽真空并用氮气置换3次,再用乙烯置换2次。冷却到30℃以后,依次加入催化剂C1(2.7mg,2.5μmol)的甲苯溶液27.4mL,再加入甲基铝氧烷(1.96mol/L,2.6ml)使Al/Zr=1000,最后加入70mL甲苯。立即将乙烯的压力升高至10大气压,在30℃下剧烈搅拌30分钟。将反应液倒出来,加入5%的盐酸乙醇溶液中和,有白色固体析出。过滤后真空干燥3小时。得到产品1.91克,聚合活性:7.6×104g·mol(Zr)-1·h-1,聚合物的熔点:135.9℃,分子量:5.49×105g·mol-1,分子量分布:10.2。
实施例3
1.催化剂C1的制备同实施例1。
2.乙烯聚合:将装有机械搅拌和热电耦的250毫升的不锈钢釜抽真空并用氮气置换3次,再用乙烯置换2次。冷却到30℃以后,依次加入催化剂C1(2.7mg,2.5μmol)的甲苯溶液27.4mL,再加入甲基铝氧烷(1.96mol/L,2.6ml)使Al/Zr=1000,最后加入70mL甲苯。立即将乙烯的压力升高至10大气压,在60℃下剧烈搅拌30分钟。将反应液倒出来,加入5%的盐酸乙醇溶液中和,有白色固体析出。过滤后真空干燥3小时。得到产品3.65克,聚合活性:1.46×105g·mol(Zr)-1·h-1,聚合物的熔点:133.7℃,分子量:9.58×106g·mol-1,分子量分布:13.6。
实施例4
1.催化剂C1的制备同实施例1。
2.乙烯聚合:将装有机械搅拌和热电耦的250毫升的不锈钢釜抽真空并用氮气置换3次,再用乙烯置换2次。冷却到30℃以后,依次加入催化剂C1(2.7mg,2.5μmol)的甲苯溶液27.4mL,再加入甲基铝氧烷(1.96mol/L,2.6ml)使Al/Zr=1000,最后加入70mL甲苯。立即将乙烯的压力升高至10大气压,在70℃下剧烈搅拌30分钟。将反应液倒出来,加入5%的盐酸乙醇溶液中和,有白色固体析出。过滤后真空干燥3小时。得到产品4.1克,聚合活性:6.41×105g·mol(Zr)-1·h-1,聚合物的熔点:133.6℃,分子量:4.65×105g·mol-1,分子量分布:9.0。
实施例5
1.催化剂C2的制备,原料中2,6-二乙基-4-甲基-N-苯乙烯基苯胺替换为2,6-二乙基-N-苯乙烯基苯胺,其它同实施例1。
2.乙烯聚合:将装有机械搅拌和热电耦的250毫升的不锈钢釜抽真空并用氮气置换3次,再用乙烯置换2次。冷却到30℃以后,依次加入催化剂C2(2.67mg,2.5μmol)的甲苯溶液37.4mL,再加入甲基铝氧烷(1.96mol/L,2.6ml)使Al/Zr=1000,最后加入60mL甲苯。立即将乙烯的压力升高至10大气压,在30℃下剧烈搅拌30分钟。将反应液倒出来,加入5%的盐酸乙醇溶液中和,有白色固体析出。过滤后真空干燥3小时。得到产品2.01克,聚合活性:8.1×104g·mol(Zr)-1·h-1,聚合物的熔点:136.6℃,分子量:3.02×105g·mol-1,分子量分布:13.0。
实施例6
1.催化剂C2的制备,原料中2,6-二乙基-4-甲基-N-苯乙烯基苯胺替换为2,6-二乙基-N-苯乙烯基苯胺,其它同实施例1。
2.乙烯聚合:将装有机械搅拌和热电耦的250毫升的不锈钢釜抽真空并用氮气置换3次,再用乙烯置换2次。冷却到30℃以后,依次加入催化剂C2(2.67mg,2.5μmol)的甲苯溶液37.4mL,再加入甲基铝氧烷(1.96mol/L,2.6ml)使Al/Zr=1000,最后加入60mL甲苯。立即将乙烯的压力升高至10大气压,在60℃下剧烈搅拌30分钟。将反应液倒出来,加入5%的盐酸乙醇溶液中和,有白色固体析出。过滤后真空干燥3小时。得到产品3.78克,聚合活性:1.51×105g·mol(Zr)-1·h-1,聚合物的熔点:133.6℃,分子量:7.96×106g·mol-1,分子量分布:15.4。
实施例7
1.催化剂C2的制备,原料中2,6-二乙基-4-甲基-N-苯乙烯基苯胺替换为2,6-二乙基-N-苯乙烯基苯胺,其它同实施例1。
2.乙烯聚合:将装有机械搅拌和热电耦的250毫升的不锈钢釜抽真空并用氮气置换3次,再用乙烯置换2次。冷却到30℃以后,依次加入催化剂C2(2.67mg,2.5μmol)的甲苯溶液37.4mL,再加入甲基铝氧烷(1.96mol/L,2.6ml)使Al/Zr=1000,最后加入60mL甲苯。立即将乙烯的压力升高至10大气压,在70℃下剧烈搅拌30分钟。将反应液倒出来,加入5%的盐酸乙醇溶液中和,有白色固体析出。过滤后真空干燥3小时。得到产品4.30克,聚合活性:7.23×105g·mol(Zr)-1·h-1,聚合物的熔点:133.4℃,分子量:5.43×105g·mol-1,分子量分布:7.80。
氧桥双核烯丙基硅桥氨基锆催化剂及其制备方法和应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。















动态评分
0.0