










专利摘要
本发明涉及一种腺苷钴胺晶型及其制备方法与应用,采用Cu‑Kα靶辐射,其X‑射线粉末衍射在衍射角2θ=5.1±0.1,6.3±0.1,7.0±0.1,7.6±0.1,8.0±0.1,8.6±0.1,9.4±0.1,13.2±0.1,13.8±0.1,15.0±0.1,17.5±0.1,19.1±0.1,21.4±0.1,23.5±0.1,25.3±0.1处有特征峰。本发明采用潜溶剂溶解腺苷钴胺粗品;采用反溶剂重结晶法得到腺苷钴胺精制品;制备过程降低杂质效果明显;操作简便,能耗低,产品纯度高于99.5%,总杂小于0.5%,收率大于98.0%,产品对光、热、高湿的稳定性显著增强;可工业化生产。
权利要求
1.一种腺苷钴胺晶型,其特征是单晶结构为单斜晶系P2
2.如权利要求1所述的晶型,其特征是DSC图谱分析结果显示,在210±2℃出现特征吸热峰。
3.如权利要求1所述的腺苷钴胺晶型的制备方法,其特征在于,采用潜溶剂-反溶剂重结晶法,步骤如下:
(1)采用潜溶剂溶解腺苷钴胺粗品;
红灯照明条件下;室温,将腺苷钴胺粗品加入到5-7倍质量的水中,搅拌5-30分钟至糊状,保持搅拌,向上述糊状物中加入潜溶剂,观察到10min内,腺苷钴胺完全溶解,得到澄清溶液;
(2)采用反溶剂重结晶法得到腺苷钴胺精制品;
红灯照明条件下;向上述溶液中加入反溶剂,得到悬浮液,室温,静置过夜,抽滤,异丙醇洗涤,在20~25℃,真空干燥3~8小时,得到腺苷钴胺晶体;
所述的步骤(1)中的潜溶剂为水与溶剂丙酮或乙腈的混合溶剂;水:丙酮的体积比=1:0.3~0.5或水:乙腈的体积比=1:0.3~0.5;
所述的步骤(2)中的反溶剂为异丙醇,正丙醇或叔丁醇,反溶剂一次性加入体系中,加入的量为步骤(1)中溶解腺苷钴胺时加入的水的体积的1~5倍。
4.如权利要求3所述的方法,其特征是所述的潜溶剂为水和丙酮;水:丙酮体积比为1:0.4。
5.如权利要求4所述的方法,其特征是所述的反溶剂为异丙醇;加入的量为步骤(1)中溶解腺苷钴胺时加入的水与异丙醇的体积比1:2.6。
说明书
技术领域
本发明涉及一种腺苷钴胺晶型及其制备方法与应用,属于结晶技术领域。
背景技术
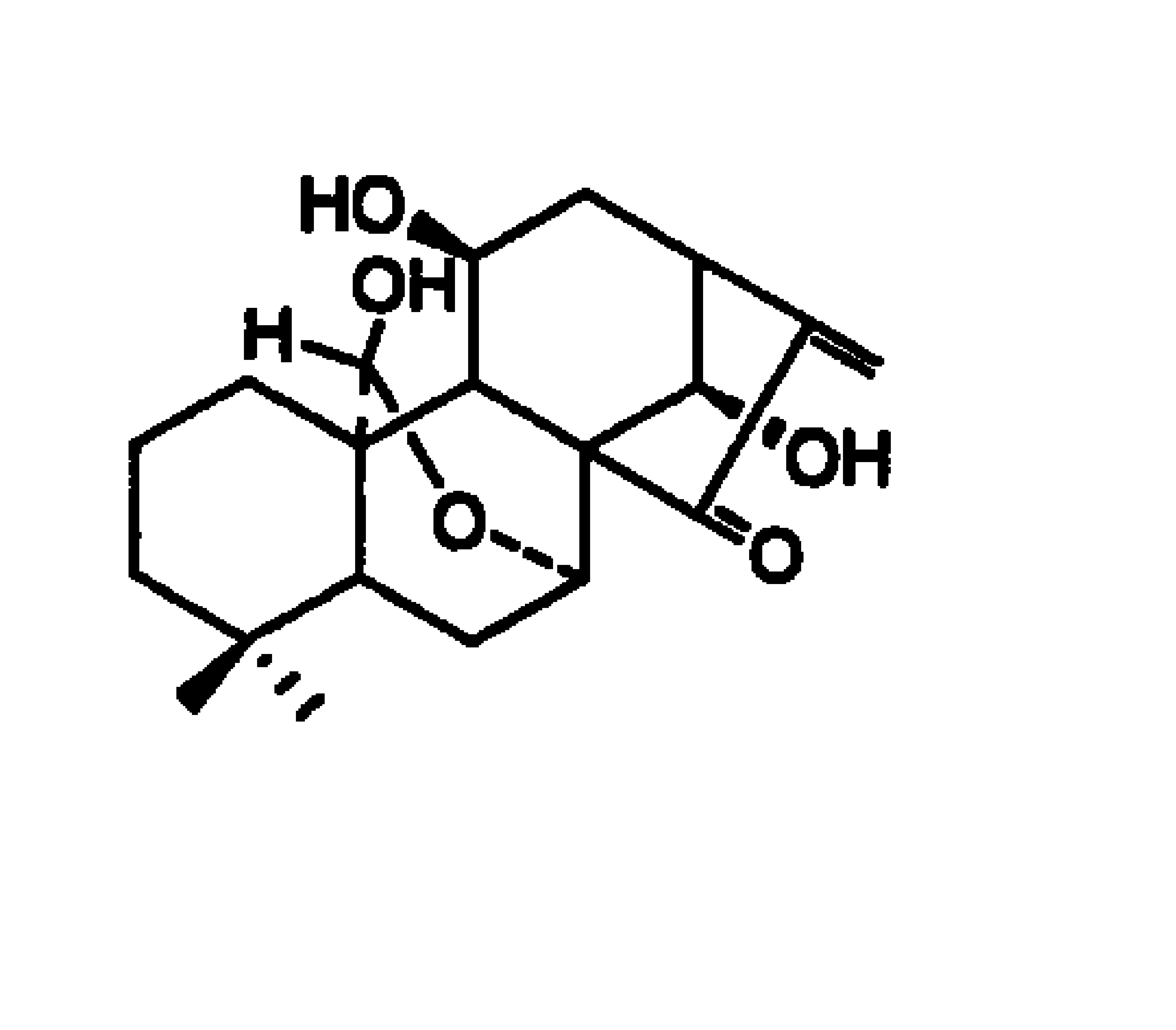
腺苷钴胺(Cobamamide也称为Adocbl或Coenzyme B12,又称脱氧腺苷钴胺素),为暗红色结晶或粉末,极具引湿性,见光极易分解。略溶于水,在乙醇中几乎不溶,在丙酮或乙醚中不溶。其化学名称为5,6-二甲基苯并咪唑基-5`-脱氧腺嘌呤核苷基钴胺,分子式为C72H100CoN18O17P,结构式如式1所示。
目前,腺苷钴胺在国际上用作生化试剂和实验试剂,只在我国用作药品,已应用多年,腺苷钴胺原料药和片剂收载于中国药典2010版和2015版。腺苷钴胺早期主要用于治疗恶性贫血,亦可与叶酸合用,用于治疗各种巨幼细胞贫血,抗叶酸药引起的贫血等。近年来,腺苷钴胺适应范围不断扩展,被用于包括神经根炎,多发性神经炎、末梢神经炎、眼肌麻痹,肝病,癌症,慢性萎缩性胃炎,生殖系统疾病治疗等领域,在临床应用过程中展现了显著的治疗效果(①刘澜波.临床合理用药杂志,2017,10(2C):43-44.②鄂瑞芳.现代药物与临床,2017,32(1):46-50.),是极具前景的多种疾病的治疗药物。
腺苷钴胺在制备和存储过程中,易产生氰钴胺、羟钴胺等杂质(①杨洪淼.中国生化药物杂志,2015,35(6):157-164.②晏敏红.海峡药学,2013,25(8):62-65),这些杂质与腺苷钴胺结构类似,同属于钴胺素类化合物。钴胺素类化合物易于形成类质同晶物(GruberGK.Vitamin B12 and B12-Proteins,Wiley-VCH,Wein-heim,1998,p335ff.),也就是说杂质易于与腺苷钴胺共同结晶析出而难以除去。目前,有关腺苷钴胺的精制方法多采用树脂吸附的方法,而通过重结晶过程除去腺苷钴胺杂质具有极大的技术难度。
我国药典2015版列出的腺苷钴胺原料药的要求是杂质含量不得高于1%,该杂质含量用于注射剂制备时,明显偏高,在我国对药品质量评价体系不断完善、评价标准不断提高的大背景下,寻找能够降低杂质的、操作简便的、符合绿色化学理念的精制方法具有重要的价值。
围绕着降低腺苷钴胺杂质的目的,我们考察了不同的重结晶条件对降低杂质的影响,研究过程中发现了新的腺苷钴胺晶型,而且,发现该晶型的制备条件具有有效降低杂质的作用。
腺苷钴胺具强的引湿性,因此,其与水分子的结合情况是需要首先考虑的问题。文献调研(①Savage H.Acta Crystallographica,1984,40(a1):C24-C24.②Bouquiere JP.Acta Crystallographica,1993,49(1):79-89.③Bouquiere J P.ActaCrystallographica,Section B:structure science,1994,B50(5):566-578.)证实,从丙酮-水混合溶剂中析出的腺苷钴胺以水合物的形式存在,1分子腺苷钴胺可结合17个水分子,这些水分子以两种形式存在,一种形式是有序水,这部分水分子有序排列于溶剂口袋中,另一种形式是无序水,这部分水分子无序排列于溶剂通道中(图1所示)。两种结合形式的水分子与腺苷钴胺的化学键的结合方式不同,因此,腺苷钴胺存在着分两次脱水的化学结构基础。通过实验和TG-DSC检测,发现腺苷钴胺在100℃以下脱去无序水,200℃以上脱去有序水,随后发生完全分解(图例可参看附图5)。
有关腺苷钴胺晶体结构及晶型的研究现状如下:
文献(Mattern CFT.Journal of Biological Chemistry,1960,235(2):489-491)报道了腺苷钴胺的单晶结构,该检测用的单晶的制备条件为:4℃,为含87.5%(体积)丙酮的水溶液。由于在X-射线下,光敏感的腺苷钴胺发生部分分解,因此所获得的数据需要较多的数学处理,获得的单晶为正交晶系P212121。然后,文献(Ouyang L.Inorganic Chemistry2004,43,1235-1241)重新确定了从水与丙酮混合溶剂中得到的腺苷钴胺的单晶结构,其晶体为正交晶系P212121,但是,该文献未提供粉末衍射数据。我们重复该实验过程获得的腺苷钴胺晶体,其粉末衍射(P-XRD)图谱见图2,该晶体的X-射线粉末衍射在衍射角2θ=6.9±0.1,7.9±0.1,9.8±0.1,13.2±0.1,14.3±0.1,16.2±0.1,18.1±0.1,19.0±0.1,20.1±0.1,21.2±0.1处有特征峰。为叙述方便,我们将其定义为A晶型腺苷钴胺。
专利JP47022716公布于1972年,该专利公布了在17℃以上和17℃以下水中重结晶获得的腺苷钴胺的2个晶型。该专利提供了两个晶型和丙酮-水混合体系中获得的晶型的X-粉末衍射图谱,但是,图谱质量较差,仅能看出较强峰的特征。从该专利附图中可以看出,丙酮-水中结晶所获得结晶的粉末衍射主峰2θ=6.9(强度100),与晶型A最强峰一致,17℃以上结晶获得的晶型的粉末衍射主峰2θ=9.1,17.0,17℃以下结晶获得的晶型的粉末衍射主峰2θ=6.6,13.0。两个从单一溶剂水中析出的晶型的粉末衍射特征峰与本发明区别明显,且未涉及降低杂质的作用,故不再赘述。
专利ZL104130303有关腺苷钴胺化合物及其药物组合物,提及一种从异丙醇、吡啶和乙醚的混合溶剂中获得的腺苷钴胺晶体,该专利说明书附上了粉末衍射图谱,同样采用Cu-Kα靶辐射,其衍射角2θ=6.1±0.1,11.2±0.1,17.0±0.1,19.1±0.1,21.0±0.1,22.2±0.1,23.5±0.1处有特征峰。为叙述方便,我们将其定义为B晶型腺苷钴胺。
综上所述,目前,已经报道了从混合溶剂中析出了两种腺苷钴胺晶型,晶型A从丙酮-水混合溶剂中析出,该晶型为人们认识已久,但是对光、热、酸碱均不稳定。晶型B的稳定性较晶型A得到了提高,但其制备过程(ZL104130303)存在着三个不足之处:
(1)采用吡啶和乙醚做溶剂,吡啶气味浓烈,有强烈刺激性,能麻醉中枢神经系统,高浓度吸入后,对人体产生严重危害,且吡啶沸点较高,为115℃,难以除去,会带来溶剂残留问题。另一溶剂乙醚极易挥发,存放时易产生过氧化物,使用时易导致操作人员产生麻醉症状,安全隐患问题严重。
(2)制备过程需要3~6分钟降温至-15~-10℃,在-15℃静置10~12小时。需要降温和低温操作。
(3)在重复该专利方法时,发现腺苷钴胺原料并未完全溶解,实验过程几乎无降低杂质的作用。
杂质的研究及控制是确保药品安全的关键要素之一,是药品研发中尤其是注射剂研发过程中风险控制意识的重要体现,目前,我国2015版药典规定的腺苷钴胺原料药杂质含量不超过1%,市售腺苷钴胺的杂质含量普遍大于0.7%,存着在通过进一步的精制,达到降低杂质的空间。我们通过重结晶技术的研究,获得了一种以较高收率获得新的晶型且进一步降低腺苷钴胺杂质的方法,目前,尚未有类似方法的报道。
发明内容
为了解决现有技术的问题,本发明的第一个目的提供一种腺苷钴胺新晶型,所获得的新晶型稳定性得到提高,更有利于产品的存贮和降低后续的制剂难度。
本发明的第二个目的是在于提供一种有较高收率的、条件温和的新晶型的制备方法,该制备方法可有效降低腺苷钴胺原料药杂质含量,可用于腺苷钴胺的精制。
本发明所述的制备方法获得了一种新的腺苷钴胺晶型,使用Cu-Kα靶辐射,其单晶衍射图谱见图3,为单斜晶系P21。C晶型与文献报道的单晶构型相比,为不同的晶系,其晶体数据对比见表1。
本发明所述的制备方法获得了一种腺苷钴胺晶型,采用Cu-Kα靶辐射,其X-射线粉末衍射在衍射角2θ=5.1±0.1,6.3±0.1,7.0±0.1,7.6±0.1,8.0±0.1,8.6±0.1,9.4±0.1,13.2±0.1,13.8±0.1,15.0±0.1,17.5±0.1,19.1±0.1,21.4±0.1,23.5±0.1,25.3±0.1处有特征峰,如图4所示。命名为C晶型腺苷钴胺。
文献报道的A,B晶型和本发明制备的晶型的X-粉末衍射的2θ角度和相对强度对比汇总于表2。从表中可以看出,三种晶型的2θ角度和相对强度的区别明显,可以判断为不同晶型。
本发明的腺苷钴胺晶型(C晶型)的制备方法,其步骤如下:
(1)采用潜溶剂溶解腺苷钴胺粗品;
红灯照明条件下;室温,将腺苷钴胺粗品加入到5-7倍质量的水中,搅拌5-30分钟至糊状,保持搅拌,向上述糊状物中加入潜溶剂,观察到10min内,腺苷钴胺完全溶解,得到澄清溶液;
(2)采用反溶剂重结晶法得到腺苷钴胺精制品;
红灯照明条件下;向上述溶液中加入反溶剂,得到悬浮液,室温,静置过夜,抽滤,异丙醇洗涤,在20~25℃,真空干燥3~8小时,得到腺苷钴胺晶型;我们命名为C晶型。
所述的步骤(1)中的潜溶剂为水与溶剂丙酮或乙腈的混合溶剂,优选丙酮。水与丙酮或乙腈的体积比为1:0.3~0.5;优选体积比为水:丙酮=1:0.4。
所述的步骤(2)中的反溶剂为异丙醇或叔丁醇,优选为异丙醇。反溶剂一次性加入体系中,加入的量为步骤(1)中溶解腺苷钴胺时加入的水的体积的1~5倍;优选为水:异丙醇=1:2.6。
本发明腺苷钴胺精制过程的溶解与析出状态,取样拍照见图15。
本发明的制备方法所用的原料为市售的腺苷钴胺,其P-XRD图谱与A晶型腺苷钴胺一致(图2),其TG-DSC热分析图谱见图5,电镜(SEM)图谱见图6,含量(HPLC)图谱见图7,红外图谱(IR)见图8。从TG-DSC图谱可以看出,A晶型腺苷钴胺在206±2℃出现一个特征放热峰,并且,100℃以下,发生无序水的脱去伴随失重约12%,在206℃发生有序水的脱去,随后发生分解。从SEM可以看出,用来做原料的市售腺苷钴胺粒度大小不一,外观呈不规则片状。按照中国药典2015版检测方法,HPLC检测主成分含量为99.24%,总杂质为0.76%。
本发明所制备得到的C晶型腺苷钴胺,其TG-DSC图谱如图9所示,SEM图谱见图10,红外图谱(IR)见图11,HPLC图谱见图12。其DSC图谱分析结果显示,在210±2℃出现一个特征吸热峰。在100℃以下发生无序水的脱去并伴随失重约12%。从SEM可以看出,本发明所制备的腺苷钴胺粒度均匀,外观呈规则块状。按照中国药典2015版检测方法,HPLC检测主成分含量为99.51%,总杂质为0.49%。
我国2015版药典要求在60℃,减压干燥至恒重时,腺苷钴胺原料药干燥失重不得超过12%,实验证实在该条件下,脱去的为无序水(或其它溶剂),因此,腺苷钴胺原料药可失去的无序水(或其它溶剂)含量应为12%以下。需要进一步说明的一个问题是,腺苷钴胺以水合物或溶剂化物形式存在,不同无序水(或其它溶剂)含量的C晶型腺苷钴胺是否具有相同的粉末衍射特征?通过改变干燥温度和时间,获得多个不同无序水(或其它溶剂)含量的C晶型腺苷钴胺样品,进行TG-DSC和X-粉末衍射分析,发现本发明方法所制得的的C晶型腺苷钴胺其粉末衍射特征与含水(或其它溶剂)量无关。为方便说明,选择有代表性的三个不同含水(或其它溶剂)量的C晶型腺苷钴胺样品a,b,c附图说明。图13为三个样品的TG-DSC分析图谱,图14为三个样品的X-粉末衍射图谱。从图13-14可以看出,三个C晶型腺苷钴胺样品在100℃以下,分别失重5.2%,7.4%和9.3%,但均在210±2℃出现特征吸热峰,三个样品的P-XRD在2θ角度和相对强度方面完全一致。以上研究证实,本方法制备的腺苷钴胺C晶型的无序水(或其它溶剂)不在晶胞中占位,对其P-XRD特征峰无影响。也就是说,本发明所制备的腺苷钴胺晶型,随着干燥条件的变化,当无序水含量发生变化时,其晶型的的X-粉末衍射特征不发生变化。
本发明所述的腺苷钴胺的精制方法为采用潜溶剂-反溶剂重结晶法。所谓潜溶剂是指药物在混合溶剂中的溶解度一般是各单一溶剂溶解度的相加平均值,在混合溶剂中各溶剂在某一比例时,药物的溶解度比在各单纯溶剂中溶解度出现极大值,这种现象称为潜溶(cosolvency),这种溶剂称为潜溶剂(cosolvent)。
选用市售的A晶型腺苷钴胺,通过大量实验研究发现,难溶性药物腺苷钴胺在水-丙酮体系,或者水-乙腈体系可形成潜溶,表3列出了腺苷钴胺在这两种体系中的溶解度(中国药典2015版溶解度测定方法)。从表中可以看出,腺苷钴胺在水中略溶,不溶于丙酮或乙腈中,但是,当水-丙酮体系,或者水-乙腈体系的体积比达到1:0.3~0.4时,此时,腺苷钴胺溶解度比在单一水溶剂中溶解度增大10倍以上。
采用潜溶原理,在少量的水-丙酮混合溶剂中,可以溶解大量的腺苷钴胺,再结合异丙醇作为反溶剂的操作,可快速析出腺苷钴胺C晶型,收率大于98%,规模可实现单批次精制腺苷钴胺50克,达到了工业生产的级别。
经过实验验证,获得的腺苷钴胺C晶型溶解于以上所述的潜溶剂后,以丙酮为反溶剂时,可析出A晶型,即以丙酮为反溶剂时,C晶型会转化为与市售品相同的A晶型,但该过程未发生杂质含量的变化,只发生了晶型的转化。也就是说,当必须使用A晶型为原料药时,可通过C晶型的制备达到降低杂质的目的,然后将C晶型转化为A晶型。
本发明方法具有以下有益效果:
潜溶剂-反溶剂重结晶法用于难溶性药物腺苷钴胺精制时,可用少量低毒性溶剂、温和的反应条件和大于98%的收率获得较高纯度的腺苷钴胺精制品。本发明所制备的腺苷钴胺C晶型粒度均匀,外观呈规则块状,按照中国药典2015版的检测方法进行含量分析,得到的主成分含量大于99.5%,总杂质小于0.5%,显著优于精制前,为解决钴胺素类化合物存在的类质同晶问题,提供了可选择的有效方法。
本发明所获得的腺苷钴胺C晶型,单晶结构及X-粉末衍射特征未见文献报道。本发明所制备的腺苷钴胺C晶型稳定性研究数据见表4-6,从实验结果可以看出,5小时以内,市售品的A晶型发生迅速降解,而自制品C晶型仅发生轻微的含量下降,可见C晶型稳定性较商品化的A晶型明显提高;C晶型腺苷钴胺对高温、高湿的稳定性也显著高于A晶型腺苷钴胺。
本发明所获得的腺苷钴胺C晶型,溶解度与A晶型相近,A晶型溶解时采用的潜溶剂的条件对C晶型完全适用。也就是本发明的制备条件可用于腺苷钴胺的二次重结晶,实验发现,二次结晶具有进一步降低杂质的作用。
表1 A、C晶型的单晶数据
表2腺苷钴胺三种晶型的X-粉末衍射对比
表3腺苷钴胺原料在水-丙酮、水-乙腈中的溶解度(25℃±2℃)
附图说明
图1腺苷钴胺水合物水分子分布图;
图2 A晶型腺苷钴胺粉末衍射图;
图3 C晶型腺苷钴胺单晶衍射图;
图4 C晶型腺苷钴胺X-粉末衍射图;
图5 A晶型腺苷钴胺TG-DSC图;
图6 A晶型腺苷钴胺SEM图(放大1000倍);
图7 A晶型腺苷钴胺HPLC图;
图8 A晶型腺苷钴胺IR图;
图9 C晶型腺苷钴胺TG-DSC图;
图10 C晶型腺苷钴胺SEM图(放大1000倍);
图11 C晶型腺苷钴胺IR图;
图12 C晶型腺苷钴胺HPLC图;
图13a无序水含量为5.2%的C晶型腺苷钴胺的TG-DSC图;
图13b无序水含量为7.4%的C晶型腺苷钴胺的TG-DSC图;
图13c无序水含量为9.3%的C晶型腺苷钴胺的TG-DSC图;
图14不同无序水含量的C晶型腺苷钴胺的X-粉末衍射图;
图15腺苷钴胺精制过程的溶解与析出图示。
具体实施方式
下面结合具体实施例对本发明作进一步的说明。
本发明制备及检测晶型所用的仪器的结构及性能如下:
红灯为5W康帅照明LED红灯球泡,厦门康帅电子有限公司。
X-射线单晶衍射分析采用Rigaku XtalAB PRO MM007 X射线衍射仪,Cu-Kα靶,在150K下收集数据。
X-射线粉末衍射分析采用Bruker D8 Advance型X射线衍射仪,Cu-Kα靶,镍单色器,工作电压40kV,工作电流40mA,步长0.02o。
TGA-DSC分析,采用美国TA公司Q600热重分析仪,升温速率10K/min。N2保护以20ml/min的速率通过DSC小室。
SEM分析,冷场发射扫描电子显微镜,德国ZEISS公司,型号ZEISS SUPRA 55,分辨率:1.0nm(15kV)/1.7nm(1kV),加速电压:0.1KV-30kV。
IR分析,红外色谱仪,赛默飞世尔科技(中国)有限公司,型号5225 Verona RdMadison,WI.53711。KBr压片。
HPLC分析,Agilent公司,型号1260,色谱柱为Intertsustain C18,检测方法为中国药典2015版第二部,腺苷钴胺。
实验例1:
红灯照明条件下,室温。将市售腺苷钴胺1g加入到7ml水中,搅拌5分钟至糊状,保持搅拌,向上述糊状物中加入3.5ml丙酮,1分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入35ml异丙醇,同时搅拌5min,搅拌速率控制在400r/min,此时体系浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用异丙醇洗涤(3*10ml),25℃真空干燥7小时,得到C晶型腺苷钴胺0.982g,收率98.2%。
所获得C晶型腺苷钴胺的X-粉末衍射图谱与图4一致,TG-DSC图谱与图9一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.5%,总杂质为0.5%。
实验例2:
红灯照明条件下,室温。将市售腺苷钴胺10g加入到60ml水中,搅拌10分钟至糊状,保持搅拌,向上述糊状物中加入24ml丙酮,3分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入156ml异丙醇,同时搅拌5min,搅拌速率控制在300r/min,此时体系浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用异丙醇洗涤(3*20ml),25℃真空干燥7小时,得到C晶型腺苷钴胺9.930g,收率99.3%。
所获得C晶型腺苷钴胺的X-粉末衍射图谱与图4一致,TG-DSC图谱与图9一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.6%,总杂质为0.5%。
实验例3:
红灯照明条件下,室温。将市售腺苷钴胺50g加入到250ml水中,搅拌30分钟至糊状,保持搅拌,向上述糊状物中加入75ml丙酮,10分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入250ml异丙醇,同时搅拌1min,搅拌速率控制在100r/min,此时体系逐渐浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用异丙醇洗涤(3*50ml),25℃真空干燥7小时,得到C晶型腺苷钴胺49.200g,收率98.4%。
该条件下所获的晶体粒度可达0.2mm,可用于X-单晶衍射检测,其单晶衍射图谱见图3,所获得C晶型腺苷钴胺的X-粉末衍射图谱与图9一致,TG-DSC图谱与图5一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.5%,总杂质为0.5%。
将本实验例所获得的C晶型腺苷钴胺进行稳定性研究,实验结果见表4-6所示,从实验结果可以看出,5小时以内,市售品的A晶型发生迅速降解,而自制品C晶型仅发生轻微的含量下降,可见C晶型稳定性较商品化的A晶型明显提高;C晶型腺苷钴胺对高温、高湿的稳定性也显著高于A晶型腺苷钴胺。
表4腺苷钴胺光照实验结果对比
注:样品分摊至培养皿中,厚度约为4mm,置于5000Lx的光橱中,按照实验方案取样后,采用中国药典2015版第二部腺苷钴胺检测方法检测含量。
表5腺苷钴胺高温实验结果对比
注:样品分摊至培养皿中,厚度约为4mm,置于60℃恒温干燥箱中,按照实验方案取样后,采用中国药典2015版第二部腺苷钴胺检测方法检测含量。
表6腺苷钴胺高湿实验结果对比
注:样品分摊至培养皿中,厚度约为4mm,置于高湿(相对湿度75%)条件下,按照实验方案取样后,采用中国药典2015版第二部腺苷钴胺检测方法检测含量。
实验例4:
红灯照明条件下,室温。将市售腺苷钴胺2g加入到14ml水中,搅拌10分钟至糊状,保持搅拌,向上述糊状物中加入6ml乙腈,3分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入35ml异丙醇,同时搅拌5min,搅拌速率控制在600r/min,此时体系浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用异丙醇洗涤(3*15ml),25℃真空干燥8小时,得到C晶型腺苷钴胺1.966g,收率98.3%。
所获得C晶型腺苷钴胺的X-粉末衍射图谱与图4一致,TG-DSC图谱与图9一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.6%,总杂质为0.5%。
实验例5:
红灯照明条件下,室温。将市售腺苷钴胺5g加入到36ml水中,搅拌16分钟至糊状,保持搅拌,向上述糊状物中加入15ml乙腈,10分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入85ml正丙醇,同时搅拌5min,搅拌速率控制在500r/min,此时体系浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用异丙醇洗涤(3*25ml),25℃真空干燥7小时,得到C晶型腺苷钴胺4.901g,收率98.0%。
所获得C晶型腺苷钴胺的X-粉末衍射图谱与图4一致,TG-DSC图谱与图9一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.5%,总杂质为0.5%。
实验例6:
红灯照明条件下,室温。将实验例2制备的C晶型腺苷钴胺1g加入到7ml水中,搅拌15分钟至糊状,保持搅拌,向上述糊状物中加入2.6ml乙腈,5分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入25ml丙酮,同时搅拌5min,搅拌速率控制在500r/min,此时体系浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用乙腈洗涤(3*10ml),25℃真空干燥7小时,得到A晶型腺苷钴胺0.990g,收率99.0%。
所获得A晶型腺苷钴胺的X-粉末衍射图谱与图2一致,TG-DSC图谱与图5一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.6%,总杂质为0.5%。
实验例7:
红灯照明条件下,室温。将实验例2制备的C晶型腺苷钴胺1g加入到7ml水中,搅拌25分钟至糊状,保持搅拌,向上述糊状物中加入3ml丙酮,5分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入25ml丙酮,同时搅拌5min,搅拌速率控制在500r/min,此时体系浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用丙酮洗涤(3*10ml),25℃真空干燥7小时,得到A晶型腺苷钴胺0.991g,收率99.1%。
所获得A晶型腺苷钴胺的X-粉末衍射图谱与图2一致,TG-DSC图谱与图5一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.6%,总杂质为0.5%。
实验例8:
红灯照明条件下,室温。将实验例2制备的C晶型腺苷钴胺1g加入到7ml水中,搅拌25分钟至糊状,保持搅拌,向上述糊状物中加入2.6ml乙腈,5分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入19ml异丙醇,同时搅拌5min,搅拌速率控制在500r/min,此时体系浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用异丙醇洗涤(3*10ml),25℃真空干燥7小时,得到C晶型腺苷钴胺0.996g,收率99.6%。
所获得C晶型腺苷钴胺的X-粉末衍射图谱与图4一致,TG-DSC图谱与图9一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.7%,总杂质为0.4%。
实验例9:
红灯照明条件下,室温。将实验例2制备的C晶型腺苷钴胺1g加入到7ml水中,搅拌25分钟至糊状,保持搅拌,向上述糊状物中加入3ml丙酮,5分钟内,腺苷钴胺完全溶解,得到澄清溶液。向上述溶液中加入18ml异丙醇,同时搅拌5min,搅拌速率控制在600r/min,此时体系浑浊。然后,室温静置过夜,抽滤析出的晶体,滤饼用异丙醇洗涤(3*10ml),25℃真空干燥7小时,得到C晶型腺苷钴胺0.993g,收率99.3%。
所获得C晶型腺苷钴胺的X-粉末衍射图谱与图4一致,TG-DSC图谱与图9一致。按照中国药典2015版的检测方法进行含量分析,得到的腺苷钴胺含量为99.7%,总杂质为0.4%。
本发明公开和提出的腺苷钴胺晶型及其制备方法与应用,本领域技术人员可通过借鉴本文内容,适当改变条件路线等环节实现,尽管本发明的方法和制备技术已通过较佳实施例子进行了描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和技术路线进行改动或重新组合,来实现最终的制备技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,他们都被视为包括在本发明精神、范围和内容中。
一种腺苷钴胺晶型及其制备方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。
















动态评分
0.0