








专利摘要
本发明涉及一种坎利酮衍生物类甾体化合物,其制备方法和其在医药领域中的用途,具体而言,涉及7α-硝基甲基-11α,17β-二羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯(式2所示的化合物)、其制备方法以及其在制备依普利酮中的用途。本发明的关键步骤在于以硝基甲烷作为亲核试剂,立体选择性在C-7位引入α-硝基甲基,从而进一步构建依普利酮C-7α位构型的羧酸甲酯结构,具有步骤简短,条件温和,成本低廉的特点。
权利要求
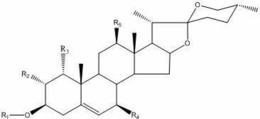
1.式2所示的化合物,
2.根据权利要求1所述的化合物的制备方法,该方法按如下步骤进行:
在碱性条件以及相转移催化剂的催化作用下,使硝基甲烷与式1所示的化合物发生1,6-Micheal加成反应,得到式2所示的化合物,
3.根据权利要求2所述的方法,其中,在所述步骤中:
使用的碱为碳酸锂、碳酸钾、碳酸钠、碳酸铯、磷酸钾、磷酸钠、氟化钠、氟化钾、氟化铯、叔丁醇钠、乙醇钠、甲醇钠、甲醇钾、乙醇钾、叔丁醇钾、2,6-二叔丁基-4-甲基-苯氧基锂或碱性氧化铝;
反应温度为25~300℃;
所述相转移催化剂为季铵盐(R1R2R3R4N+)mXm-,其中,m为1、2或3;Xm-为F-、Cl-、Br-、I-、NO3-、PO43-、CO32-或SO42-;′
R1、R2、R3和R4各自独立地为C1-C30直链或支链烷基;或-(CH2)nR5,其中n=0-10的整数,R5为未取代的或C1-C6烷基取代的C6-C15芳香基;
或者R1为苄基、苯基乙基或苯基丙基,并且R2、R3和R4与氮原子形成被1~3个取代基取代的1-氮杂二环[2.2.2]辛烷的环状结构,所述取代基为C1-C3烷基、乙烯基或1’-羟基-1’-(喹啉-4”-基)甲基;
使用的溶剂为选自N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜、六甲基磷酰胺、硝基苯、吡啶、乙二醇二甲醚、乙二醇二乙醚、1,4-二氧六环中的一种或者两种以上的混合溶剂。
4.根据权利要求3所述的方法,其中,
所述碱为碳酸钾、碳酸钠、碳酸锂、碳酸铯、磷酸锂、磷酸钾或磷酸钠;
所述反应温度为50~150℃;
R1、R2、R3和R4各自独立地为C1-C18直链或支链烷基;或-(CH2)nR5,其中n=0-10的整数,R5为未取代的或C1-C3烷基取代的苯基;
或者R1为苄基、苯基乙基或苯基丙基,并且R2、R3和R4与氮原子形成被1~3个取代基取代的1-氮杂二环[2.2.2]辛烷的环状结构,所述取代基为C1-C3烷基、乙烯基或1’-羟基-1’-(喹啉-4”-基)甲基;
所述溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜或六甲基磷酰胺。
5.根据权利要求2-4中任一项所述的方法,其中,所述相转移催化剂为N-苄基氯化辛可宁、N-苄基氯化辛可宁丁、十八烷基三甲基氯化铵、苄基二甲基十八烷基氯化铵或苄基三乙基氯化铵。
6.根据权利要求1所述的化合物在制备依普利酮中的用途,其中,按照如下步骤制备依普利酮:
1)将式2所示的化合物溶于溶剂中,在氧化剂的作用下,通过Nef反应将式2中的C7α-硝基甲基转化为C7α-羧基,得到式3所示的化合物;
2)使式3所示的化合物在碱性条件下与甲酯化试剂发生甲酯化反应生成式4所示的化合物;
3)使式4所示的化合物在碱性条件下与酰卤试剂反应发生酯化反应得到式5所示的化合物;
其中,R为甲基磺酰基、对甲苯磺酰基、苯磺酰基、乙酰基或三氟乙酰基;
4)在碱性条件下,式5所示的化合物在脱水剂的作用下发生消除反应得到式6所示的化合物;
5)使式6所示的化合物的C9(11)位烯烃在环氧化试剂的作用下发生环氧反应,得到式7所示的化合物,即,依普利酮;
7.根据权利要求6所述的用途,其中,
在步骤1)中:
反应温度为0~80℃;
所述溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜、六甲基磷酰胺、叔丁醇或甲醇;
所述氧化剂为亚硝酸钠/醋酸复合试剂;高锰酸钾;过硫酸氢钾;四丙基高钌酸铵/N-甲基吗啉复合试剂;醋酸铜/氧气复合试剂或双(三甲基硅)过氧化合物;
在步骤2)中:
使用的溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜、六甲基磷酰胺、丙酮或水/丙酮复合溶剂;
所述甲酯化试剂为碘甲烷、硫酸二甲酯或重氮甲烷;
所述碱为碳酸钾、碳酸钠、碳酸锂、碳酸氢钾或碳酸氢钠;
在步骤3)中:
所述酰卤试剂为甲基磺酰氯、对甲苯磺酰氯、苯磺酰氯、乙酰氯或三氟乙酸酐;
使用的碱为三乙胺或吡啶;
使用的溶剂为吡啶、二氯甲烷或氯仿;
反应温度为-25~40℃;
在步骤4)中:
反应温度为40~150℃;
使用的碱为醋酸钾、醋酸钠、甲酸钾或甲酸钠;
所述脱水剂为酸酐;
使用的溶剂为甲酸、乙酸或三氟醋酸;
在步骤5)中:
反应溶液pH值≥7;
所述环氧化试剂为间氯过苯甲酸、双氧水/三氯乙腈复合试剂、双氧水/三氯乙酸酐复合试剂或双氧水/三氯乙酰胺复合试剂;
使用的溶剂为CH2Cl2、CHCl3、CCl4或乙酸乙酯。
8.根据权利要求7所述的用途,其中,
在步骤1)中:
反应温度为0~60℃;
在步骤4)中:
所述脱水剂为乙酸酐或三氟醋酐;
所述反应温度为80~100℃;
在步骤5)中:
反应溶液pH=8~9。
说明书
技术领域
本发明涉及一种坎利酮衍生物类甾体化合物,其制备方法和其在医药领域中的用途,具体而言,涉及7α-硝基甲基-11α,17β-二羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯(式2所示的化合物)、其制备方法以及其在制备依普利酮(如式7所示的化合物)中的用途。
背景技术
依普利酮是新型且具有选择性特点的醛固酮拮抗剂,可与醛固酮直接结合从而抑制其作用,作用与螺内酯近似,长期服用对性腺不良反应轻微。依普利酮(Eplerenone),其结构如下面的式7所示,1984年由瑞士Ciba-Geigy Ag公司制备,并于2002年由美国FDA批准上市。
依普利酮是主要用于治疗原发性高血压以及心肌梗塞后的心力衰竭的药物,也是第一个获准上市的选择性醛固酮受体阻断剂。在体内由CYP450 3A4代谢,半衰期为4~6小时。依普利酮可用于提高急性心脏病发作后的充血性心力衰竭患者的生存率。
依普利酮的合成工作,国内外已有相关专利和文献报道,早期合成工作主要由瑞士Ciba-Geigy Ag公司的Grob J.小组完成(EP122232、US4559332)。之后,G.D.Searle公司的John NG小组(WO9721720)、Pharmacia & Upjohn的Wuts小组(US2004087562-A1)、White小组(US2007066579-A1)以及Pearlman小组(WO2003082895)分别开发了不同的合成路线。近年也有许多国内专利公开做过相关的报道(CN101121652、CN1724557和CN1749266),合成的关键部分都是集中在C-7位α构型的羧酸甲酯结构单元的构建,但这些方法中需要用到剧毒的氰化物试剂,如氰化钠、氰基丙酮或二乙基氰基铝;同时氰基转换要用到价格昂贵的DIBAH试剂;或者在引入C-7α位羰基时需要用到贵金属,如醋酸钯等;或者立体选择性不佳;或者需要在低温(-46℃)条件下用臭氧氧化呋喃环等苛刻反应条件和繁琐的操作步骤。
本发明的关键步骤在于以硝基甲烷作为亲核试剂,立体选择性在C-7位引入α-硝基甲基,从而进一步构建依普利酮C-7位α构型的羧酸甲酯结构,具有步骤简短,条件温和,成本低廉的特点。
发明内容
技术问题
为了解决现有技术的上述问题,本发明提供了用于制备依普利酮的重要中间体化合物、该中间体化合物的制备方法及该中间体化合物在制备依普利酮中的用途。本发明提供的依普利酮的新的制备方法克服了以往路线中需要低温、使用贵重试剂或者剧毒试剂缺点,并且该合成路线以工业上易得的11α-羟基坎利酮为起始原料,经过共5步,以55%的总收率得到目标产物依普立酮,所涉及的关键步骤的反应都是立体专一性的,反应条件温和、易于放大生产,因而降低了生产成本,在工业生产中具有很高的应用价值。
技术方案
因此,本发明的目的在于提供式2所示的化合物(即7α-硝基甲基-11α,17β-二羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯)。
本发明的另一目的在于提供式2所示的化合物的制备方法。
本发明的又一目的在于提供式2所示的化合物在制备式7所示的化合物(即依普利酮)中的用途。
根据本发明的一个方面,其提供了式2所示的化合物,
根据本发明的另一方面,提供了式2所示的化合物的制备方法。
本发明所述的式2所示的化合物的制备方法,该方法按如下步骤进行:
在碱性条件以及相转移催化剂的催化作用下,使硝基甲烷与式1所示的化合物,即11α-羟基坎利酮发生1,6-Micheal加成反应,立体专一性地得到C-7α硝基甲基的加成产物,即式2所示的化合物(7α-硝基甲基-11α,17β-二羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯),该加成产物可不经分离直接用于下一步;
具体为将碱、硝基甲烷、相转移催化剂和式1所示的化合物混于溶剂中,反应得到式2所示的化合物;
其中所述碱为碳酸锂、碳酸钾、碳酸钠、碳酸铯、磷酸钾、磷酸钠、氟化钠、氟化钾、氟化铯、叔丁醇钠(NaOt-Bu)、乙醇钠(NaOEt)、甲醇钠(NaOMe)、甲醇钾(KOMe)、乙醇钾(KOEt)、叔丁醇钾(KOt-Bu)、2,6-二叔丁基-4-甲基-苯氧基锂或碱性氧化铝,优选为碳酸钾、碳酸钠、碳酸锂、碳酸铯、磷酸锂、磷酸钾或磷酸钠;
其中所述相转移催化剂为季铵盐(R1R2R3R4N+)mXm-,其中,m为1、2或3;Xm-可以为F-、Cl-、Br-、I-、NO3-、PO43-、CO32-或SO42-;
R1、R2、R3和R4各自独立地为C1-C30直链或支链烷基,优选C1-C18直链或支链烷基;或-(CH2)nR5,其中n=0-10的整数,R5为未取代的或被C1-C6烷基取代的C6-C15芳香基,优选为未取代或被C1-C3烷基取代的苯基,进一步地,-(CH2)nR5优选为苄基;
或者,R1为苄基、苯基乙基或苯基丙基,并且R2、R3和R4与氮原子形成被1~3个取代基取代的1-氮杂二环[2.2.2]辛烷的环状结构,所述取代基为C1-C3烷基、乙烯基或1’-羟基-1’-(喹啉-4”-基)甲基。
所述季铵盐优选为N-苄基氯化辛可宁、N-苄基氯化辛可宁丁、十八烷基三甲基氯化铵、苄基二甲基十八烷基氯化铵或苄基三乙基氯化铵;优选为苄基二甲基十八烷基氯化铵或苄基三乙基氯化铵;
所述的溶剂为选自N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMAC)、二甲亚砜(DMSO)、六甲基磷酰胺(HMPA)、硝基苯、吡啶、乙二醇二甲醚、乙二醇二乙醚、1,4-二氧六环中的一种或者两种以上的混合溶剂,优选为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜或六甲基磷酰胺;
所述反应温度为25~300℃;优选为50~150℃。
本发明的另一方面提供了式2所示的化合物在制备依普利酮中的用途,其中,按如下步骤进行:
1)将式2所示的化合物溶于溶剂中,在氧化剂的作用下,通过Nef反应将式2中的C7α-硝基甲基转化为C7α-羧基,得到如式3所示的化合物,即7α-羧酸-11α,17β-二羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯,该化合物可直接用于下一步;
具体为将式2所示的化合物溶于溶剂中,在0~80℃的反应温度下加入氧化剂,发生Nef反应将硝基甲基转化成羧基即得如式3所示的化合物;
所述溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜、六甲基磷酰胺、叔丁醇或甲醇;
所述反应温度优选为0~60℃;
所述氧化剂为亚硝酸钠/醋酸(AcOH)复合试剂、高锰酸钾(pH值为10~11)、过硫酸氢钾(Oxone)(pH值为10~11)、四丙基高钌酸铵(TPAP)/N-甲基吗啉(NMO)复合试剂、醋酸铜(Cu(OAc)2)/氧气(O2)复合试剂或双(三甲基硅)过氧化合物(bis(trimethylsilyl)peroxide);
2)使式3所示的化合物在碱性条件下与甲酯化试剂发生甲酯化反应生成式4所示的化合物,即7α-羧酸甲酯-11α,17β-二羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯,该化合物可直接用于下一步;
具体为将式3所示的化合物溶解于溶剂中,加入碱和甲酯化试剂,发生甲酯化反应生成如式4所示的化合物,该化合物可直接用于下一步;
所述溶剂为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜、六甲基磷酰胺、丙酮或水/丙酮复合溶剂;
所述甲酯化试剂为碘甲烷、硫酸二甲酯或重氮甲烷;
所述碱为碳酸钾、碳酸钠、碳酸锂、碳酸氢钾或碳酸氢钠;
3)使式4所示的化合物,即7α-羧酸甲酯-11α,17β-二羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯中的C11-羟基在碱性条件下与酰卤试剂发生酯化反应,得到如式5所示的化合物,即7α-羧酸甲酯-11α-R氧基-17β-羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯,式5所示的化合物可不经分离直接用于下一步反应;
其中,R为甲基磺酰基、对甲苯磺酰基、苯磺酰基、乙酰基或三氟乙酰基;
具体为将式4所示的化合物溶于溶剂中,然后加入碱和酰卤试剂反应得到式5所示的化合物;
所述酰卤试剂为甲基磺酰氯、对甲苯磺酰氯、苯磺酰氯、乙酰氯或三氟乙酸酐;
所述碱为三乙胺或吡啶;
所述溶剂为吡啶、二氯甲烷或氯仿;
所述反应温度为-25~40℃;
4)在碱性条件下,式5所示的化合物,即7α-羧酸甲酯-11α-R-氧基-17β-羟基-3-氧代-17α-孕甾-4-烯-21-羧酸-γ-内酯,在脱水剂的作用下发生消除反应得到式6所示的化合物,即7α-羧酸甲酯-17β-羟基-3-氧代-17α-孕甾-4,9(11)-二烯-21-羧酸-γ-内酯;
具体为将碱、脱水剂在室温下溶解于溶剂中,再加入式5所示的化合物在40~150℃的反应温度下,发生消除反应形成C9(11)烯双键,得到式6所示的化合物;
其中所述碱为醋酸钾、醋酸钠、甲酸钾或甲酸钠;
所述脱水剂为酸酐,优选为乙酸酐或三氟醋酐;
所述溶剂为甲酸、乙酸或三氟醋酸;
所述反应温度优选为80~100℃;
5)使式6所示的化合物的C9(11)位烯烃在环氧化试剂的作用下发生环氧反应,得到式7所示的化合物,即依普利酮;
具体为将式6所示的化合物溶解于溶剂中,调节反应溶液pH值≥7,再加入环氧化试剂,充分搅拌并反应得到式7所示的化合物;
其中所述环氧化试剂为间氯过苯甲酸(m-CPBA)、双氧水/三氯乙腈(H2O2/Cl3CCN)复合试剂、双氧水/三氯乙酸酐(H2O2/(Cl3CCO)2O)复合试剂或双氧水/三氯乙酰胺(H2O2/CCl3CONH2)复合试剂;
其中所述溶剂为CH2Cl2、CHCl3、CCl4或乙酸乙酯;
以及反应溶液优选pH=8~9。
具体实施方式
下面结合实施例对本发明进行进一步阐述,其并不用于限制本发明。
下述实施例中,熔点由Buchi 510熔点仪测定,核磁共振由Bruker AMX-400型和Varian型核磁共振仪测定,TMS为内标,化学位移单位为ppm;质谱和高分辨由Finnigan MAT-95型高分辨质谱仪测定;柱层析用硅胶200-300目,青岛海洋化工厂生产;TLC硅胶板为烟台化工厂生产的HSGF-254型薄层层析预制板。N-苄基氯化辛可宁根据文献Tetrahedron 56(26):4447-4451制备;N-苄基氯化辛可宁丁和苄基二甲基十八烷基氯化铵分别从梯希爱化成工业发展有限公司(Tciamerica)购买;十八烷基三甲基氯化铵和苄基三乙基氯化铵分别从国药集团化学试剂有限公司购买。
式1所示的化合物,即11α-羟基坎利酮由浙江仙居神州药业有限公司提供。
实施例1:式2所示的化合物的制备
向接有氯化钙干燥管和空气冷凝管的两颈瓶中加入式1所示的化合物(20g,56mmol)、季铵盐(6mmol)(见表1)、硝基甲烷(60ml,112mmol)和190ml N,N-二甲基甲酰胺(DMF),溶解完全后升温至90~100℃,边搅拌边加入K2CO3(78g,565mmol),保持充分搅拌,并在此温度下反应48小时。反应终止时,先降温,等温度完全降低至室温后,垫硅藻土过滤,并用二氯甲烷(100ml×3)洗滤饼,合并二氯甲烷层。加入少许10%(w/w)的稀盐酸调节有机层pH值至6。先减压旋干所有溶剂得到棕色泡沫状固体,再将该固体溶于二氯甲烷溶液中(500ml)并过滤,二氯甲烷有机层再用稀盐酸(250ml×3)洗,稀盐酸水层需用二氯甲烷(50ml×3)再洗一遍,合并二氯甲烷有机层。再用饱和食盐水(250ml×3)洗涤。得到的二氯甲烷有机层用无水硫酸钠干燥,旋干,得到棕色粗品23g,可不经分离直接用于下一步。乙酸乙酯重结晶得到式2所示的化合物的纯品。
表-1:所述的季铵盐
mp:281-282℃
51°(c=0.24,CHCl3)
IR vmax(KBr):3467,2950,1766,1662,1550,1458,1441,1419,1382,1344,1274,1192。
UVmax:242nm
1H NMR(300MHz,CDCl3)δ5.82(s,1H),4.38-4.20(m,2H),4.10(tt,J=10.29,10.29,4.95,4.95Hz,1H),2.81(td,J=13.99,4.16,4.16Hz,1H),2.73-2.27(m,10H),2.07-1.70(m,7H),1.68-1.59(m,2H),1.50(ddd,J=24.57,12.19,5.69Hz,1H),1.38(s,3H),1.04(s,3H)。
13C NMR(100MHz,CDCl3)δ199.092,176.239,164.980,128.330,94.707,74.029,68.710,53.387,46.162,45.170,43.219,39.867,37.704,37.471,36.477,35.951,35.328,34.207,31.127,29.087,22.276,18.343,15.679。
EI MS(70eV,m/z):417(M+,21%),399(17%),370(44%),111(100%)。
HR-MS(EI):理论计算C23H31NO6(M+)417.2151,结果417.2153。
元素分析:理论计算C23H31NO6:C,66.17;H,7.48;N,3.35.结果:C,66.35;H,7.63;N,3.31。
实施例2:式4所示的化合物的制备
1)将式2所示的化合物(5g,0.012mol)溶于40ml N,N-二甲基甲酰胺(DMF)中,之后升温至45℃。向该溶液中加入醋酸(2.9ml,0.051mol),充分搅拌后,在30min内分批加入NaNO2(2.484g,0.036mol),在加完NaNO2后,再让其反应一小时,之后,先将醋酸(2.9ml,0.051mol)和3ml DMF混合,再将其逐滴滴加入上述反应液中,完全滴完后,约需4小时左右,TLC分析原料完全消失,之后降温至0℃,再加入饱和亚硫酸钠溶液(54ml,0.089mol)淬灭反应再升温至室温,过夜彻底终止反应。反应完全终止后,减压旋干所有溶剂,得到粉末状固体再用丙酮溶解,并过滤,浓缩滤液至100ml,即得式3所示的化合物的粗品丙酮溶液,直接用于下一步。
2)将上述式3所示的化合物的粗品丙酮溶液降温至0℃,之后依次加入碘甲烷(1.5ml,0.024mol)和碳酸钾(K2CO3)(5g,0.036mol)并充分搅拌后,升至室温,反应过夜,TLC检测原料完全消失即可。过滤除去K2CO3,并用二氯甲烷(100ml×3)洗滤饼,合并有机层,旋干。再用二氯甲烷250ml溶解,后依次用稀盐酸(10%w/w)、饱和NaHCO3溶液、饱和硫代硫酸钠溶液、饱和食盐水洗,再用无水硫酸钠干燥、过滤、旋干得到式4所示的化合物的粗品,并直接用于下一步。式4所示的化合物的粗品用二氯甲烷/甲苯重结晶得到无色透明晶体(4g,收率:80%)。
mp.:230-232℃
1H NMR(300MHz,CDCl3)δ5.68(s,1H),4.04(dt,J=10.23,9.84,3.98Hz,1H),3.64(s,3H),2.85-2.71(m,2H),2.68-2.18(m,8H),2.10-1.57(m,9H),1.49-1.40(m,2H),1.35(s,3H),1.00(s,3H)。
HR-MS(EI)calcd for C24H32O6(M+)416.221 found 416.225
元素分析:理论计算C24H32O6:C,69.21;H,7.74.结果:C,68.32;H,7.72
UVmax:244nm
实施例3:式6所示的化合物的制备(R=甲基磺酰基)
1)在0℃下,将式4所示的化合物(4g,9.6mmol)溶于54ml二氯甲烷溶液中,再加入甲基磺酰氯(1.4ml,17mmol),随后,先将三乙胺(4.65ml,31mmol)和2ml二氯甲烷混合,再滴加到上述反应液中,完全滴加完后,升温至室温反应3小时,TLC检测,当式4所示的化合物完全消失后,反应即完全。所有溶剂全部旋干,得到式5-1所示的化合物的粗品,该产物为白色固体(微红),直接用于下一步。
2)在氮气保护、室温下,将乙酸酐(1.8ml)逐滴滴加到甲酸钾(1.3g,0.015mol)的甲酸(3.6ml)溶液中,搅拌并升温至70℃反应17小时,之后向反应液中加入式5-1所示的化合物,并升温至100℃,反应2小时后,TLC检测原料完全消失,向其中加入冰块使其内部温度迅速降低到50℃以下,减压旋干所有溶剂得到白色固体,二氯甲烷溶解后用饱和NaHCO3洗、稀盐酸洗、饱和食盐水洗、旋干后得到式6所示的化合物粗品,并可以直接用于下一步。乙酸乙酯/石油醚重结晶得到纯品(3.05g,收率80%)。
mp.:204-206℃
1H NMR(300MHz,CDCl3)δ5.67(s,1H),5.60(d,J=5.84Hz,1H),3.54(s,3H),2.93(dd,J=4.48,2.00Hz,1H),2.78(ddd,J=14.93,5.15,1.64Hz,1H),2.61-2.36(m,6H),2.33-2.05(m,5H),1.99-1.75(m,5H),1.51-1.38(m,2H),1.35(s,3H),0.90(s,3H)。
13C NMR(100MHz,CDCl3)δ198.805,176.667,172.828,166.858,142.504,125.871,119.134,95.252,51.605,44.638,43.935,43.250,40.719,40.526,35.902,35.586,34.359,33.912,33.068,31.616,29.378,27.354,23.448,14.234。
实施例4:式7所示的化合物的制备
在0℃下,依次加入式6所示的化合物(3.05g,7.5mmol)、K2HPO4(5.5g,24mmol)、CH2Cl2(65ml)、Cl3CCONH2(4g,36mmol)之后边搅拌边加入双氧水(21ml,30% w/w,260mmol.)加入到单口瓶中,反应溶液pH值为9,再升温至10-15℃,充分搅拌并反应24小时,原料完全反应,加入饱和Na2S2O3水溶液(75ml)终止反应,再用CH2Cl2萃取,合并有机层,旋干,丙酮重结晶得到化合物式7所示的化合物(无色透明晶体)(3.06g,收率98%)。
+2.2°(c 0.415,CHCl3)
UVmax:240nm
元素分析:理论计算:C24H30O6:C,69.54;H,7.30.结果:C,69.25;H,7.30;
mp.:242-244℃
1H NMR(300MHz,CDCl3)δ1.01(s,3H),1.48(s,3H),5.89(s,1H),3.64(s,3H),3.11(d,J=5.16Hz,1H),2.87(dd,J=7.30,4.19)。
13C NMR(100MHz,CDCl3)δ198.004,176.245,172.583,164.991,127.085,94.568,65.324,51.606,51.490,43.858,41.289,39.732,38.757,37.274,34.978,34.841,33.085,31.02 5,30.950,28.951,26.982,22.319,22.149,16.225。
EI-MS(70eV,m/z):414(M+),399([M-Me]+),396([M-H2O]+),355([M-COOMe]+)。
一种坎利酮衍生物类甾体化合物、其制备方法及其在制备依普利酮中的用途专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。


















动态评分
0.0