








专利摘要
本发明公开了一种14C标记的雌激素硫酸盐共轭物及其制备方法,属于放射性同位素14C标记的化合物领域。本发明的一种14C标记的雌激素硫酸盐共轭物的制备方法,在雌激素硫酸盐共轭物的结构中引入14C放射性同位素标记,14C标记位点在C-3。放射性自显影-多功能扫描成像系统测试结果表明,放射性纯度达到98%,合成的14C标记化合物为研究雌激素共轭物在环境中的归趋提供了条件。
权利要求
1.一种14C标记的雌激素硫酸盐共轭物,其特征在于,在雌激素硫酸盐共轭物的结构中引入14C放射性同位素标记,它的结构通式为:
其中,*为14C标记位点,上述结构式含C的编号。
2.根据权利要求1所述的14C标记的雌激素硫酸盐共轭物,其特征在于,在所述的共轭物中,-R1为-SO3-或-H;-R2为=O、 或
3.根据权利要求1或2所述的14C标记的雌激素硫酸盐共轭物,其特征在于,在所述的共轭物中,R1、R2的对应关系如下:
4.一种权利要求1至3任意一项所述的14C标记的雌激素硫酸盐共轭物的制备方法,其步骤为:
(A)将溶于乙酸乙酯的5mCi/mmol、放射性纯度99%的[3-14C]-雌酮(1)或[3-14C]-17β-雌二醇(2)或[3-14C]-17α-雌二醇(2)’加入梨形瓶中;
(B)缓慢氮气流除去乙酸乙酯后,加入三氧化硫-三乙胺复合物,再加入200μL无水吡啶,室温下或90-95℃搅拌反应3h,得到中间产物;
(C)缓慢氮气流除去吡啶,加入1M的KOH甲醇溶液至不再有白色沉淀产生,转移到离心管中,离心,吸取上清液;
(D)向沉淀中继续加入甲醇,漩涡震荡提取,离心,吸取上清液,这一提取过程重复4-6次;
(E)合并步骤(C)、(D)中离心管中多次吸取的上清液,旋转蒸发浓缩,用制备薄层色谱分离提纯;硅胶制备板为20cm×20cm×2mm,展开剂为氯仿、甲醇和氨水的混合液;
(F)通过放射性自显影,利用多功能扫描成像系统,根据其中的Rf值确定产物位置,将含有产物的硅胶板区域刮下,得到的硅胶粉末用甲醇提取5-6次,合并提取液,旋转蒸发浓缩,得到最终产物,计算产率;
(G)产物经薄层色谱分析,硅胶分析板为3cm×10cm×0.25mm,展开剂为氯仿、甲醇、氨水的混合液,通过放射性自显影,利用多功能扫描成像系统,测得放射性纯度。
说明书
技术领域
本发明涉及放射性同位素14C标记的化合物领域,特别涉及环境中雌激素硫酸盐共轭物及其制备方法。
背景技术
环境雌激素又称环境内分泌干扰物,按来源分为:天然类固醇雌激素、人工合成雌激素、植物雌激素、真菌性雌激素、环境化学污染物等。其中,人体和动物排出的天然类固醇雌激素雌酮(E1)、17β-雌二醇(17β-E2),以及人工合成的口服避孕药17α-乙炔基雌二醇(17α-EE2)在水环境中的雌激素活性最高,尤其17β-E2在浓度低至ng/L级就会产生内分泌干扰作用。目前研究发现类固醇雌激素以引起生物效应的浓度存在于污水、泉水、河流、海洋、沉积物和土壤中。由于肠道细菌的作用,少量类固醇雌激素以自由态的形式从动物粪便中排出,大部分类固醇雌激素以极性的葡萄糖醛酸盐或硫酸盐共轭物的形式从尿液排出。雌激素共轭物不能与雌激素受体结合,活性较小,但在环境介质中会通过水解重新释放具有雌激素活性的自由态雌激素,具有潜在的环境风险,Isobe等研究指出雌酮-3-硫酸盐(E1-3S)可以引起日本鹌鹑睾丸细胞减少以及睾丸重量下降,认为E1-3S是一种干扰野生动物内分泌系统的潜在因素。有文献报道,在污水处理系统中,部分雌激素葡萄糖醛酸共轭物可以转化为硫酸盐共轭物。也有文献报道,在下水道污泥中,雌激素共轭物较之自由态雌激素的降解更为缓慢,雌激素的硫酸盐共轭物比葡萄糖醛酸盐共轭物更持久,因此,研究雌激素硫酸盐共轭物在环境中的归趋具有重要意义。
但研究雌激素共轭物在土壤、底泥、畜禽粪便中的分布、迁移、转化、降解、代谢和残留等环境行为时,由于基质的干扰,往往需要对样品进行复杂的前处理,不仅耗费大量的时间,而且由于处理步骤的繁复,往往会导致回收率差。另外,由于GC-MS或LC-MS等仪器检测限的限制,实验中使用的浓度往往高于环境中的实际浓度,因此很难反映雌激素共轭物在环境中归趋的真实情况。采用14C放射性同位素示踪技术,不仅可以准确跟踪复杂体系(如天然环境样品)中的雌激素共轭物,而且可以准确定位降解代谢产物和残留物在体系中的分布,为降解和转化过程及机理的研究提供有效手段。而且这种基于放射性的测定方法可以减少研究中使用的化学浓度,使研究更接近环境真实情况。但是14C标记化合物价格十分昂贵,且有关14C标记的雌激素共轭物的实验室合成文献鲜见。
Shrestha等曾报道经三步合成[4-14C]-17β-雌二醇-17-硫酸钾([4-14C]-17β-E2-17S钾盐)的方法:首先利用苯甲酰氯将[4-14C]-17β-雌二醇([4-14C]-17β-E2)的C3位酚羟基保护,然后在50℃条件下与三氧化硫—吡啶复合物反应,将C-17位羟基硫酸酯化,最后在NaOH甲醇液中将C3位的苯甲酰保护基水解,生成最终产物[4-14C]-17β-E2-17S。(Shrestha,S.L.;Bai,X.L.;Smith,D.J.;Hakk,H.;Casey,F.X.M.;Larsen,G.L.Padmanabhana,G.(2011)Synthesis and characterization of radiolabeled17β-estradiol conjugates.J.Labelled Compd.Radiopharm.54:267-271.)。其不足之处在于,该方法的步骤复杂且合成的成本高。
发明内容
1.要解决的技术问题
针对现有技术中,14C标记雌激素硫酸盐共轭物仅涉及[4-14C]-17β-E2-17S钾盐一个共轭物,且制备历经三步,步骤复杂,本发明提供了一种14C标记的雌激素硫酸盐共轭物及其制备方法。该方法步骤简化,合成方法具有普适性,目标化合物放射性纯度同样达到98%以上,为后续采用14C放射性同位素示踪技术研究雌激素硫酸盐共轭物的环境归趋提供了条件。
2.技术方案
发明原理:本发明的目的是建立14C标记的雌激素硫酸盐共轭物的合成方法,同位素标记位点在C3位(如图1)。原料[3-14C]-雌酮([3-14C]-E1)、[3-14C]-17β-雌二醇([3-14C]-17β-E2)和[3-14C]-17α-雌二醇([3-14C]-17α-E2)来源于实验室的前期合成。由于14C标记化合物原料得来不易,代价昂贵,所以在合成标记的雌激素硫酸盐共轭物之前,首先以非标记的雌激素合成相应的非标记雌激素硫酸盐共轭物,以确定反应条件,并用液相色谱-质谱联用仪和核磁共振仪等对合成的非标记硫酸盐共轭物进行结构表征,确证结构。
本发明的目的通过以下技术方案实现。
一种14C标记的雌激素硫酸盐共轭物,在雌激素硫酸盐共轭物的结构中引入14C放射性同位素标记,它的结构通式为:
其中,*为14C标记位点,上述结构式含C的编号。
进一步地,在所述的共轭物中,-R1为-SO3-或-H;-R2为=O、 或
进一步地,在所述的共轭物中,R1、R2的对应关系如下:
一种所述的14C标记的雌激素硫酸盐共轭物的制备方法(如图2),其步骤为:
(A)将溶于乙酸乙酯的5mCi/mmol、放射性纯度99%的[3-14C]-雌酮([3-14C]-E1)(1)或[3-14C]-17β-雌二醇([3-14C]-17β-E2)(2)或[3-14C]-17α-雌二醇([3-14C]-17α-E2)(2)’加入梨形瓶中;
(B)缓慢氮气流除去乙酸乙酯后,加入三氧化硫-三乙胺复合物((Et)3N·SO3),再加入200μL无水吡啶,室温下或90-95℃搅拌反应3h,得到中间产物;
(C)缓慢氮气流除去吡啶,加入1M的KOH甲醇溶液至不再有白色沉淀产生,转移到离心管中,离心,吸取上清液;
(D)向沉淀中继续加入甲醇,漩涡震荡提取,离心,吸取上清液,这一提取过程重复4-6次;
(E)合并步骤(C)、(D)中离心管中多次吸取的上清液,旋转蒸发浓缩,用制备薄层色谱分离提纯;硅胶制备板为20cm×20cm×2mm,展开剂为氯仿、甲醇和氨水的混合液;
(F)通过放射性自显影,利用多功能扫描成像系统,根据其中的Rf值确定产物位置,将含有产物的硅胶板区域刮下,得到的硅胶粉末用甲醇提取5-6次,合并提取液,旋转蒸发浓缩,得到最终产物,计算产率;
(G)产物经薄层色谱分析,硅胶分析板为3cm×10cm×0.25mm,展开剂为氯仿、甲醇、氨水的混合液,通过放射性自显影,利用多功能扫描成像系统,测得放射性纯度。
本发明有代表性的化合物包括:[3-14C]-雌酮-3-硫酸钾([3-14C]-E1-3S钾盐)(3)、[3-14C]-17β-雌二醇-17-硫酸钾([3-14C]-17β-E2-17S钾盐)(4)、[3-14C]-17α-雌二醇-17-硫酸钾([3-14C]-17α-E2-17S钾盐)(4)’、[3-14C]-17β-雌二醇-3,17-二硫酸钾([3-14C]-17β-E2-3,17diS钾盐)(5)、[3-14C]-17α-雌二醇-3,17-二硫酸钾([3-14C]-17α-E2-3,17diS钾盐)(5)’。
在合成14C标记共轭物时,[3-14C]-E1和[3-14C]-E2的化学浓度一般较低,为保证微量反应顺利进行,可加入一定量非标记的E1或E2,或加大三氧化硫-三乙胺的量(如10倍摩尔量)。对于[3-14C]-E1-3S钾盐,反应一般不会产生副产物。但是对于[3-14C]-E2-17S钾盐,当增加三氧化硫-三乙胺的量且反应时间过长(如过夜)时,可能会生成[3-14C]-E2-3,17diS钾盐,但是制备型薄层色谱分离提纯后,同样可以得到纯的[3-14C]-E2-17S钾盐。
在合成14C标记共轭物时,向反应体系中加入KOH甲醇溶液后会产生白色沉淀。反应生成的[3-14C]-E2-3,17diS钾盐几乎全部都在沉淀中,而[3-14C]-E1-3S钾盐和[3-14C]-E2-17S钾盐大部分留在上清液中。但为了提高产率,合成14C标记共轭物时,均应用甲醇对产生的白色沉淀进行漩涡震荡提取,提取次数通过测定放射性回收率确定,一般提取4-6次,每次1-2mL甲醇。
3.有益效果
相比于现有技术,本发明的优点在于:
(1)采用14C放射性同位素示踪技术研究环境污染物,研究雌激素硫酸盐共轭物的环境行为,可以减少前处理过程并减少实验中使用的污染物浓度,使研究更接近环境真实情况;
(2)利用14C标记的雌激素硫酸盐共轭物,结合放射性同位素示踪技术,可以准确跟踪复杂体系(如天然环境样品)中的雌激素共轭物,可以准确定位降解代谢产物和残留物在体系中的分布,为降解和转化过程及机理的研究提供有效手段;
(3)本发明建立了14C标记的雌激素硫酸盐共轭物的合成方法,与Shrestha等报道的经三步合成[4-14C]-17β-E2-17S钾盐的方法相比,本发明中[3-14C]-17β-雌二醇-17-硫酸钾([3-14C]-17β-E2-17S钾盐)(4)的合成方法更为简单;对于本方法合成的14C标记的雌激素硫酸盐共轭物,放射性自显影-多功能扫描成像系统测试结果表明,放射性纯度同样达到了98%以上;14C标记的雌激素硫酸盐共轭物的获得,使得本领域技术人员利用14C放射性同位素示踪技术,进而研究雌激素硫酸盐共轭物在环境中的归趋成为可能。
附图说明
图1为14C标记的雌激素硫酸盐共轭物的结构式(含C的编号;*为14C标记位点);
图2为[3-14C]-E1-3S钾盐(3)、[3-14C]-17β-E2-17S钾盐(4)、[3-14C]-17α-E2-17S钾盐(4)’、[3-14C]-17β-E2-3,17diS钾盐(5)和[3-14C]-17α-E2-3,17diS钾盐(5)’的合成流程图;
其中,(Et)3N·SO3为三氧化硫-三乙胺复合物,RT为室温,*为14C标记位点;
以下图3-1至3-5为本发明产物的相应质谱图,其中:
图3-1为雌酮-3-硫酸钾(E1-3S钾盐)的电喷雾质谱图;
图3-2为17β-雌二醇-17-硫酸钾(17β-E2-17S钾盐)的电喷雾质谱图;
图3-3为17β-雌二醇-3,17-二硫酸钾(17β-E2-3,17diS钾盐)的电喷雾质谱图;
图3-4为17β-雌二醇-3-硫酸钠(17β-E2-3S钠盐)标品的Q-TOF二级质谱图;
图3-5为17β-雌二醇-17-硫酸钾(17β-E2-17S钾盐)的Q-TOF二级质谱图;
图4-1为雌酮(E1)的13C NMR图;
图4-2为17β-雌二醇(17β-E2)的13C NMR图;
图4-3为雌酮-3-硫酸钾(E1-3S钾盐)的13C NMR图;
图4-4为17β-雌二醇-17-硫酸钾(17β-E2-17S钾盐)的13C NMR图;
图4-5为17β-雌二醇-3,17-二硫酸钾(17β-E2-3,17diS钾盐)的13C NMR图;
图4-6为雌酮(E1)的1H NMR图;
图4-7为17β-雌二醇(17β-E2)的1H NMR图;
图4-8为雌酮-3-硫酸钾(E1-3S钾盐)的1H NMR图;
图4-9为17β-雌二醇-17-硫酸钾(17β-E2-17S钾盐)的1H NMR图;
图4-10为17β-雌二醇-3,17-二硫酸钾(17β-E2-3,17diS钾盐)的1H NMR图。
具体实施方式
下面结合说明书附图和具体的实施例,对本发明作详细描述。
实施例1:14C标记雌激素共轭物[3-14C]-E1-3S钾盐(3)的合成
结合图1、2,14C标记雌激素共轭物[3-14C]-E1-3S钾盐(3)合成步骤为:
(A)将[3-14C]-E1(1.13mg,21μCi,5mCi/mmol,400μL乙酸乙酯)加入5mL梨形瓶中;
(B)缓慢氮气流除去乙酸乙酯后,加入9.2mg三氧化硫-三乙胺复合物(与[3-14C]-E1相比,摩尔比约为1:15.7),再加入200μL无水吡啶,室温下搅拌反应3h,生成中间产物[3-14C]-E1-3S三乙胺复合物。无水吡啶是反应溶剂介质,使得反应体系保持碱性。
(C)缓慢氮气流除去吡啶,加入1M KOH甲醇溶液至不再有白色沉淀产生,转移到离心管中,离心,吸取上清液。这一步生成的[3-14C]-E1-3S钾盐大部分在上清液中。白色沉淀为反应生成的无机盐,以及少许的未被溶解的[3-14C]-E1-3S钾盐。
(D)向沉淀中继续加入甲醇,漩涡震荡提取,离心,吸取上清液,这一提取过程重复4次。反复提取,是为了充分的将[3-14C]-E1-3S钾盐提取到溶剂中。
(E)合并步骤(C)、(D)中离心管中多次吸取的上清液,旋转蒸发浓缩,用制备薄层色谱分离提纯(硅胶制备板为20cm×20cm×2mm,展开剂为氯仿:甲醇:氨水=5:1:0.1(V:V:V),所用氨水NH3含量25.0~28.0%)。
(F)通过放射性自显影,利用多功能扫描成像系统确定产物位置(Rf值为0.21),将含有产物的硅胶板区域刮下,得到的硅胶粉末用甲醇提取5次,合并提取液,旋转蒸发浓缩,得到[3-14C]-E1-3S钾盐10μCi,产率为48%。
(G)产物经薄层色谱分析(硅胶分析板为3cm×10cm×0.25mm,展开剂为氯仿:甲醇:氨水=5:1:0.1(V:V:V),所用氨水NH3含量25.0~28.0%),通过放射性自显影-多功能扫描成像系统(Rf值0.21),测得其放射性纯度为99%。
在合成[3-14C]-E1-3S钾盐之前,首先合成相应的非标记E1-3S钾盐,以确定反应条件;并用液相色谱-质谱联用仪和核磁共振仪等对合成的E1-3S钾盐进行结构表征,以确证结构。
对应非标记雌激素共轭物E1-3S钾盐(3)的合成:将1.7g E1和1.24g三氧化硫-三乙胺复合物(摩尔比约为1:1.1)溶于数毫升无水吡啶中,室温下搅拌反应2h,旋转蒸发除去吡啶。加入1M KOH甲醇溶液,此时会产生大量白色沉淀,继续加入KOH至不再有白色沉淀产生,滤去沉淀,将滤液减压旋蒸至干,在此过程中逐渐有白色晶体析出。用无水乙醚洗涤白色晶体以除去残留的E1,最终得到2.3g E1-3S钾盐白色晶体,产率为94.1%,纯度为97.1%。
非标记雌激素共轭物E1-3S钾盐(3)的结构表征:
ESI-MS:m/z[M-K]-=349.1(100%),电喷雾质谱数据见图3-1。
13C NMR(E1(1)in DMSO-d6/TMS):δ/ppm13.954(C-18),21.589(C-15),26.030(C-11),26.597(C-7),29.524(C-6),31.832(C-12),35.805(C-16),38.436(C-8),43.882(C-9),48.62(C-13),50.039(C-14),113.248(C-2),115.432(C-4),126.461(C-1),130.041(C-10),137.744(C-5),155.531(C-3),220.209(C-17)。13C NMR数据见图4-1。
13C NMR(E1-3S potassium(3)in D2O):δ/ppm13.507(C-18),21.069(C-15),25.503(C-11),25.984(C-7),29.043(C-6),31.116(C-12),35.368(C-16),38.184(C-8),43.568(C-9),48.351(C-13),49.746(C-14),118.652(C-2),121.391(C-4),126.517(C-1),137.182(C-5),138.317(C-10),149.746(C-3),226.869(C-17)。13C NMR数据见图4-3。
由13C NMR可见,与反应物E1(1)相比,3的C-2、C-4和C-10的化学位移δ增加,向低场移动,C-3位的化学位移δ减少,向高场移动,表明在C-3位形成硫酸酯。这是因为,当苯环上C-3位上的羟基取代(推电子取代基)变成磺酸基取代(吸电子取代基)时,由于电子的共振离域,使得邻、对位碳C-2、C-4和C-10上的电荷密度减少,屏蔽减小,向低场移动。同样因为共振和离域,C-3则向高场移动。
1H NMR(E1(1)in DMSO-d6/TMS):δ/ppm0.824(3H,s,CH3-18),1.304-1.594(6H,m,H-12,H-7,H-15),1.742-1.758(1H,dd,H-14),1.900-1.962(2H,m,H-8,H-11),2.023-2.133(2H,m,H-16),2.302(1H,m,H-11),2.404-2.458(1H,m,H-9),2.744-2.801(2H,t,H-6),6.462(1H,s,H-4),6.514-6.530(1H,dd,H-2),7.056-7.040(1H,d,H-1),9.014(Ar-OH,OH-3,与溶剂DMSO-d6缔合形成分子间氢键)。1H NMR数据见图4-6。
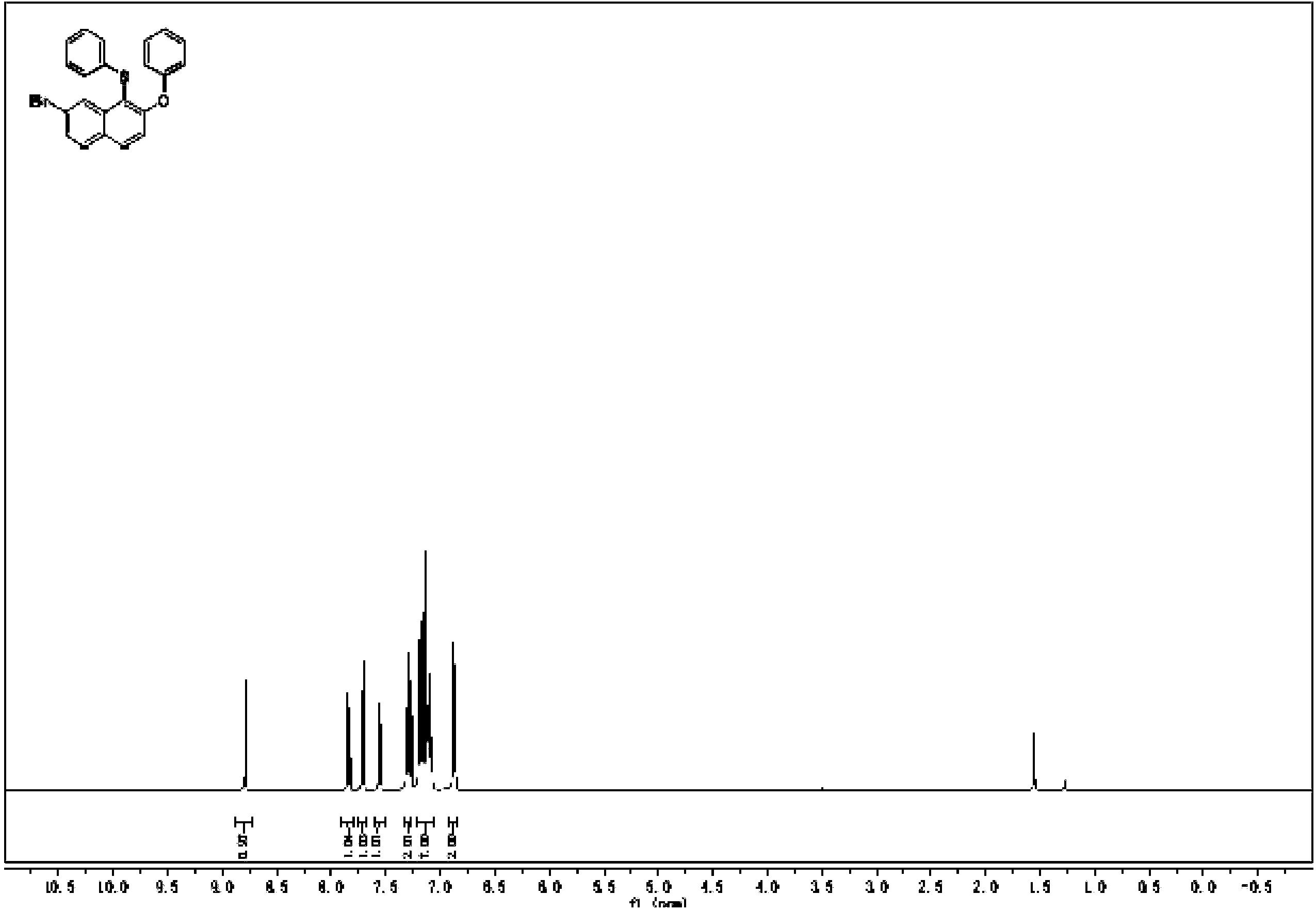
1H NMR(E1-3S potassium(3)in D2O):δ/ppm0.772(3H,s,CH3-18),1.187-1.578(6H,m,H-12,H-7,H-15),1.712-1.776(1H,dd,H-14),1.855-1.886(1H,dd,H-8),1.947-1.974(1H,m,H-11),2.051-2.155(2H,m,H-16),2.249-2.258(1H,m,H-11),2.438-2.494(1H,m,H-9),2.692-2.758(2H,t,H-6),6.950(1H,s,H-4),6.968-6.989(1H,dd,H-2),7.213-7.230(1H,d,H-1)。1H NMR数据见图4-8。
由1H NMR可见,与反应物E1(1)相比,3的C-3位的邻、间位碳上的H-1、H-2和H-4,化学位移δ增加,向低场位移,佐证了在C-3位形成硫酸酯,与13C NMR数据一致。
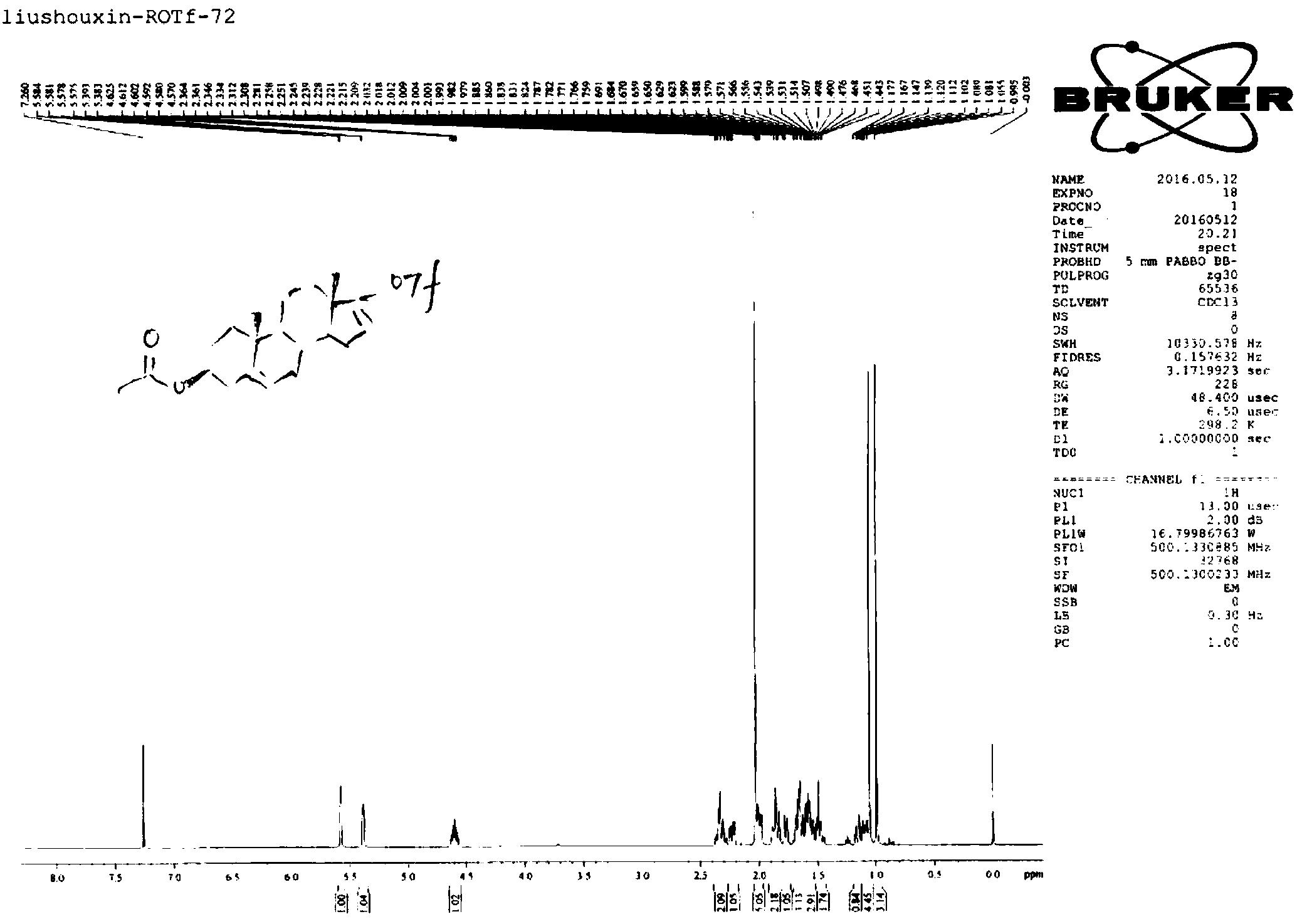
实施例2:14C标记雌激素共轭物[3-14C]-17β-E2-17S钾盐(4)的合成
结合图1、2,14C标记雌激素共轭物[3-14C]-17β-E2-17S钾盐(4)的合成步骤为:
(A)将[3-14C]-17β-E2(0.61mg,11.2μCi,5mCi/mmol,370μL乙酸乙酯)加入5mL梨形瓶中。
(B)缓慢氮气流除去乙酸乙酯后,加入2.4mg非标记的17β-E2和11.2mg三氧化硫-三乙胺复合物(与[3-14C]-17β-E2和17β-E2二者之和相比,摩尔比约为1:6),再加入200μL无水吡啶,室温下搅拌反应3h。
(C)缓慢氮气流除去吡啶,加入1M KOH甲醇溶液至不再有白色沉淀产生,转移到离心管中,离心,吸取上清液。这一步生成的[3-14C]-17β-E2-17S钾盐大部分在上清液中。白色沉淀为反应生成的无机盐,以及少许的未被溶解的[3-14C]-17β-E2-17S钾盐。
(D)向沉淀中继续加入甲醇,漩涡震荡提取,离心,吸取上清液,这一提取过程重复6次。反复提取,是为了充分的将[3-14C]-17β-E2-17S钾盐提取到溶剂中。
(E)合并步骤(C)、(D)中离心管中多次吸取的上清液,旋转蒸发浓缩,用制备薄层色谱分离提纯(硅胶制备板为20cm×20cm×2mm,展开剂为氯仿:甲醇:氨水=3:1:0.1(V:V:V),所用氨水NH3含量25.0~28.0%)。
(F)通过放射性自显影,利用多功能扫描成像系统确定产物位置(Rf值0.29),将含有产物的硅胶板区域刮下,得到的硅胶粉末用甲醇提取6次,合并提取液,旋转蒸发浓缩,得到[3-14C]-17β-E2-17S钾盐2.8μCi,产率为25%。
(G)产物经薄层色谱分析(硅胶分析板为3cm×10cm×0.25mm,展开剂为氯仿:甲醇:氨水=3:1:0.1(V:V:V),所用氨水NH3含量25.0~28.0%),通过放射性自显影-多功能扫描成像系统(Rf值0.29),测得其放射性纯度为99%。
在合成[3-14C]-17β-E2-17S钾盐之前,首先合成相应的非标记17β-E2-17S钾盐,以确定反应条件;并用液相色谱-质谱联用仪和核磁共振仪等对合成的17β-E2-17S钾盐进行结构表征,以确证结构。
对应非标记雌激素共轭物17β-E2-17S钾盐(4)的合成:2g17β-E2和1.46g三氧化硫-三乙胺复合物(摩尔比约为1:1.1)溶于数毫升无水吡啶中,室温下搅拌反应2h,旋转蒸发除去吡啶,加入1M KOH甲醇溶液,此时会产生大量白色沉淀,继续加入KOH至不再有白色沉淀产生,滤去沉淀。将滤液减压旋蒸浓缩至一定体积,加入无水乙醚,此时在滤液与无水乙醚的界面处会产生白色固体,白色固体以下的液体呈油状。将油状液体用吸管移出,剩余部分过滤,收集白色固体。向移出的油状液体中继续加入无水乙醚,收集界面处新产生的白色固体。反复多次,直至加入无水乙醚,无白色固体析出。如果界面处析出的固体颜色发黄并呈结块状,可将此黄色固体重新溶解在甲醇中,再加入无水乙醚,至界面处析出固体的颜色为白色,再过滤收集。用无水乙醚洗涤收集到的白色固体,以除去残留的17β-E2,最终得到1.1g17β-E2-17S钾盐白色固体,产率为38.4%,纯度为93.5%。
非标记雌激素共轭物17β-E2-17S钾盐(4)的结构表征:
ESI-MS:m/z[M-K]-=351.3(100%),电喷雾质谱数据见图3-2、图3-3。在17β-E2和三氧化硫—三乙胺复合物(摩尔比约为1:1.1)反应时,可能会在芳香环的C-3位上键合,也可能在脂环的C-17位上键合,分别对应形成共轭物17β-E2-3S或17β-E2-17S。为了确定产物4的结构,购得17β-E2-3S钠盐标品,然后进一步对照做了产物4与17β-E2-3S钠盐的Q-TOFLCMS二级质谱负离子扫描,见图3-4,3-5。结果显示,17β-E2-3S钠盐的二级质谱出现了m/z351.1280,271.1705,和79.9565三组峰,分别代表分子离子峰[M-Na]-,子离子[M-Na-SO3]-,离子自由基[SO3-·]。但是产物4的二级质谱则出现了m/z351.1297,96.9609,分别代表分子离子峰[M-K]-和HSO4-。据文献报道17β-E2-3S钠盐不会产生m/z97的HSO4-,原因在于芳香环C-3位上没有多余的氢原子可提供。结合13C NMR和1H NMR光谱分析,反应产物确证为4,即17β-E2-17S钾盐。17β-E2与三氧化硫-三乙胺复合物((Et)3N·SO3)在吡啶中反应,专属性地使C-17位羟基硫酸酯化,可能是因为其中反应生成的3-单硫酸酯三乙胺复合物在干燥吡啶中不稳定,很容易地分解为原料17β-E2,然后C-17位羟基硫酸酯化。
13C NMR(17β-E2(2)in DMSO-d6/TMS):δ/ppm11.701(C-18),23.25(C-15),26.569(C-11),27.429(C-7),29.637(C-6),30.375(C-16),37.060(C-12),39.162(C-8),3.264(C-13),43.978(C-9),49.984(C-14),80.569(C-17),113.156(C-2),115.391(C-4),126.418(C-1),130.892(C-10),137.547(C-5),155.346(C-3)。13C NMR数据见图4-2。
13C NMR(17β-E2-17S potassium(4)in D2O):δ/ppm11.514(C-18),22.816(C-15),26.092(C-11),26.996(C-7),27.904(C-16),29.347(C-6),36.407(C-12),38.202(C-8),42.883(C-13),43.515(C-9),48.920(C-14),87.814(C-17),114.593(C-2),116.765(C-4),126.648(C-1),129.734(C-10),138.125(C-5),158.126(C-3)。13C NMR数据见图4-4。
由13C NMR可见,与反应物17β-E2(2)相比,4的C-17向低场移动,这是由于在C-17位形成硫酸酯,硫酸酯基团电负性高于羟基,去屏蔽效应增加,所以C-17化学位移δ增加,移向低场。与反应物2相比,4的C-3位的化学位移变化较小,而且芳香环上其他C的化学位移几乎没有变化,表明硫酸酯化位置在C-17位上,而不是C-3位。
1H NMR(17β-E2(2)in DMSO-d6/TMS):δ/ppm0.667(3H,s,CH3-18),1.071-1.392(7H,m,H-12,H-7,H-15,1/2H-11),1.552-1.608(1H,q,H-11),1.755-1.785(1H,m,H-14),1.823-1.903(2H,m,H-16),2.045-2.087(1H,m,H-8),2.212-2.248(1H,m,H-9),2.672-2.728(2H,t,H-6),3.505-3.538(1H,t,H-17β),4.491(br,OH-17),6.434(1H,s,H-4),6.494-6.515(1H,d,H-2),7.034-7.051(1H,d,H-1),8.975(Ar-OH,OH-3,与溶剂DMSO-d6缔合形成分子间氢键)。1HNMR数据见图4-7。
1H NMR(17β-E2-17S potassium(4)in D2O):δ/ppm0.780(3H,s,CH3-18),1.158-1.390(6H,m,H-12,H-7,H-15),1.662-1.695(2H,q,H-11),1.786-1.813(1H,dd,H-14),1.867-1.929(1H,dd,H-8),2.055-2.160(2H,m,H-16),2.217-2.227(1H,m,H-9),2.679-2.740(2H,t,H-6),4.238-4.272(1H,t,H-17β),6.464(1H,s,H-4),6.509-6.524(1H,d,H-2),7.109-7.126(1H,d,H-1)。1H NMR数据见图4-9。
由1H NMR可见,2和4的H-17β位的化学位移δ分别为3.505-3.538和4.238-4.272,与反应物2相比,4的H-17β向低场移动,佐证了在C-17位形成硫酸酯,与13C NMR数据一致。同时,由于16位H与17位硫酸根的氧原子形成分子内氢键,去屏蔽效应增强,导致H-16化学位移δ增加,向低场移动。与反应物2相比,4的C-3位的邻、间位碳上的H-1、H-2、和H-4,化学位移δ几乎没有变化,佐证了硫酸酯化位置在C-17位上,而不是C-3位。由于4以D2O作为溶剂,故而C-3位上酚羟基的质子吸收峰未观察到。
实施例3:14C标记雌激素共轭物[3-14C]-17β-E2-3,17diS钾盐(5)的合成
结合图1、2,14C标记雌激素共轭物[3-14C]-17β-E2-3,17diS钾盐(5)的合成步骤为:
(A)将[3-14C]-17β-E2(0.013mg,0.24μCi,5mCi/mmol,230μL乙酸乙酯)加入5-mL梨形瓶中。
(B)缓慢氮气流除去乙酸乙酯后,加入3mg非标记的17β-E2和10.5mg三氧化硫-三乙胺复合物(与[3-14C]-17β-E2和17β-E2二者之和相比,摩尔比约为1:6.8),再加入200μL无水吡啶,90-95℃搅拌反应3h。
(C)缓慢氮气流除去吡啶,加入1M KOH甲醇溶液至不再有白色沉淀产生,转移到离心管中,离心,吸取上清液。这一步反应生成的[3-14C]-E2-3,17diS钾盐几乎全部都在沉淀中。
(D)向沉淀中继续加入甲醇,漩涡震荡提取,离心,吸取上清液,这一过程重复5次。反复提取,是为了充分的将[3-14C]-E2-3,17diS钾盐提取到溶剂中。
(E)合并步骤(C)、(D)中离心管中多次吸取的上清液,旋转蒸发浓缩,用制备薄层色谱分离提纯(硅胶制备板为20cm×20cm×2mm,展开剂为氯仿:甲醇:氨水=2:1:0.2(V:V:V),所用氨水NH3含量25.0~28.0%)。
(F)通过放射性自显影,利用多功能扫描成像系统确定产物位置(Rf值0.22),将含有产物的硅胶板区域刮下,得到的硅胶粉末用甲醇提取6次,合并提取液,旋转蒸发浓缩,得到[3-14C]-17β-E2-3,17diS钾盐0.13μCi,产率为54%。
(G)产物经薄层色谱分析(硅胶分析板为3cm×10cm×0.25mm,展开剂为氯仿:甲醇:氨水=2:1:0.2(V:V:V),所用氨水NH3含量25.0~28.0%),通过放射性自显影-多功能扫描成像系统(Rf值0.22),测得其放射性纯度为98%。
在合成[3-14C]-17β-E2-3,17diS钾盐之前,首先合成相应的非标记17β-E2-3,17diS钾盐,以确定反应条件;并用液相色谱-质谱联用仪和核磁共振仪等对合成的17β-E2-3,17diS钾盐进行结构表征,以确证结构。
对应非标记雌激素共轭物17β-E2-3,17diS钾盐(5)的合成:将700mg17β-E2和2.8g三氧化硫-三乙胺复合物(摩尔比约为1:7.8)溶于数毫升无水吡啶中,90-95℃搅拌反应3h,旋转蒸发除去吡啶。加入1M KOH甲醇溶液,此时会产生大量沉淀,继续加入至不再有沉淀产生,过滤收集沉淀,得淡黄色粗品17β-E2-3,17diS钾盐。将粗品溶于水后,用Waters制备色谱提纯,样品浓度200mM,流动相为乙腈:100mM乙酸铵溶液=18:82(V:V),色谱柱为Sunfire C18(19×150mm,10μm),流速为15mL/min,柱温为室温,波长215nm,进样量为1mL,收集2.3-4.3min目标组分。馏分收集液旋转蒸发除去乙腈,冻干除去水和乙酸铵,得394mg17β-E2-3,17diS钾盐白色絮状固体,产率为30.1%,纯度为98.3%。
非标记雌激素共轭物17β-E2-3,17diS钾盐(5)的结构表征:
ESI-MS:m/z[M-K]-=431.0(50%),[M-2K]2-=215.2(100%)。电喷雾质谱数据见图4-3。
13C NMR(17β-E2(2)in DMSO-d6/TMS):δ/ppm11.701(C-18),23.25(C-15),26.569(C-11),27.429(C-7),29.637(C-6),30.375(C-16),37.060(C-12),39.162(C-8),3.264(C-13),43.978(C-9),49.984(C-14),80.569(C-17),113.156(C-2),115.391(C-4),126.418(C-1),130.892(C-10),137.547(C-5),155.346(C-3)。13C NMR数据见图4-2。
13C NMR(17β-E2-3,17diS dipotassium(5)in D2O):δ/ppm11.076(C-18),22.575(C-15),25.604(C-11),26.360(C-7),27.670(C-16),28.894(C-6),36.149(C-12),37.869(C-8),42.691(C-13),43.371(C-9),48.679(C-14),88.530(C-17),118.425(C-2),121.311(C-4),126.697(C-1),138.460(C-5),138.924(C-10),148.998(C-3)。13C NMR数据见图4-5。
由13C NMR可见,与反应物17β-E2(2)相比,5的C-17向低场移动,这是由于在C-17位形成硫酸酯,硫酸酯基团电负性高于羟基,去屏蔽效应增加,所以C-17化学位移值增加,移向低场。与反应物2相比,5的C-2、C-4和C-10的化学位移增加,向低场移动,C-3位的化学位移δ减少,向高场移动,表明在C-3位形成硫酸酯。这是因为,当苯环上C-3位上的羟基取代(推电子取代基)变成磺酸基取代(吸电子取代基)时,由于电子的共振离域,使得邻、对位碳C-2、C-4和C-10上的电荷密度减少,屏蔽减小,向低场移动。同样因为共振和离域,C-3则向高场移动。
1H NMR(17β-E2(2)in DMSO-d6/TMS):δ/ppm0.667(3H,s,CH3-18),1.071-1.392(7H,m,H-12,H-7,H-15,1/2H-11),1.552-1.608(1H,q,H-11),1.755-1.785(1H,m,H-14),1.823-1.903(2H,m,H-16),2.045-2.087(1H,m,H-8),2.212-2.248(1H,m,H-9),2.672-2.728(2H,t,H-6),3.505-3.538(1H,t,H-17β),4.491(br,OH-17),6.434(1H,s,H-4),6.494-6.515(1H,d,H-2),7.034-7.051(1H,d,H-1),8.975(Ar-OH,OH-3,Ar-OH,OH-3,与溶剂DMSO-d6缔合形成分子间氢键)。1H NMR数据见图4-7。
1H NMR(17β-E2-3,17diS dipotassium(5)in D2O):δ/ppm0.750(3H,s,CH3-18),1.214-1.438(6H,m,H-12,H-7,H-15),1.634-1.724(2H,q,H-11),1.824-1.847(1H,dd,H-14),1.911-1.935(1H,dd,H-8),2.090-2.260(2H,m,H-16),2.268-2.294(1H,m,H-9),2.786-2.804(2H,t,H-6),4.248-4.281(1H,t,H-17β),6.982(1H,s,H-4),6.994-7.011(1H,d,H-2),7.318-7.335(1H,d,H-1)。1H NMR数据见图4-10。
由1H NMR可见,2和5的H-17β位的化学位移δ分别为3.505-3.538和4.248-4.281,与反应物2相比,5的H-17β向低场移动,佐证了在C-17位形成硫酸酯,与13C NMR数据一致。同时,由于16位H与17位硫酸根的氧原子形成分子内氢键,去屏蔽效应增强,导致H-16化学位移δ增加,向低场移动。与反应物2相比,5的C-3位的邻、间位碳上的H-1、H-2和H-4,化学位移δ增加,向低场位移,佐证了在C-3位形成硫酸酯,与13C NMR数据一致。
一种标记的雌激素硫酸盐共轭物及其制备方法专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。




















动态评分
0.0