

IPC分类号 : C12N1/19,C12N15/63,C12P15/00,C12R1/865







专利摘要
本发明公开了生产次丹参酮二烯的酿酒酵母基因工程菌及其构建方法与应用。本发明所提供的基因工程菌是按照包括以下步骤的方法构建的:在出发酿酒酵母中引入外源的丹参柯巴基焦磷酸合酶基因(SmCPS)和丹参次丹参酮二烯合酶基因(SmKSL)得到生产次丹参酮二烯的重组酿酒酵母,记作重组酿酒酵母ZD-T-000和ZD-T-010。在此基础上,提高3-羟基-3-甲基戊二酰辅酶A还原酶、萜类调控因子蛋白UPC2、牻牛儿牻牛儿基焦磷酸合酶、法呢基焦磷酸合酶中的一种或几种蛋白的活性,构建得到24株(ZD-T-001~ZD-T-008,ZD-T-011~ZD-T-018,ZD-T-021~ZD-T-028)高产次丹参酮二烯的重组酿酒酵母菌株。本发明所构建的酿酒酵母ZD-Tans-001在好养氧条件下,发酵6天时可生产487.91mg/L的次丹参酮二烯。 CGMCCNo.5296 2011.09.28
权利要求
1.酿酒酵母(Saccharomyces cerevisiae BY4742)ZD-Tans-001,其保藏号为CGMCC No.5296。
2.一种基因工程菌的构建方法,包括以下步骤:
在出发酿酒酵母中引入外源的柯巴基焦磷酸合酶和次丹参酮二烯合酶,得到重组酿酒酵母,记作重组酿酒酵母I。
3.根据权利要求2所述的方法,其特征在于:所述方法还包括以下步骤:
在所述重组酿酒酵母I中提高3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2的活性,得到重组酿酒酵母,记作重组酿酒酵母II。
在所述重组酿酒酵母I中提高牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶的活性,得到重组酿酒酵母,记作重组酿酒酵母III。
4.根据权利要求3所述的方法,其特征在于:所述方法还包括以下步骤:
在所述重组酿酒酵母III中提高3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2的活性,得到重组酿酒酵母,记作重组酿酒酵母IV。
5.根据权利要求2-4中任一所述的方法,其特征在于:
所述引入外源的柯巴基焦磷酸合酶和次丹参酮二烯合酶是通过转化入携带外源柯巴基焦磷酸合酶和次丹参酮二烯合酶基因的质粒,或在酿酒酵母染色体上整合外源柯巴基焦磷酸合酶和次丹参酮二烯合酶基因。
所述提高3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2的活性是通过转化入携带3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2基因的质粒,或在酿酒酵母染色体上整合3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2基因。
所述提高牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶的活性是通过转化入携带牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶基因的质粒,或在酿酒酵母染色体上整合牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶基因。
6.根据权利要求5所述的方法,其特征在于:
所述在酿酒酵母染色体上整合基因是整合在δDNA位点。
所述携带柯巴基焦磷酸合酶和次丹参酮二烯合酶基因的质粒,复制位点为2MICRON,筛选标记为尿嘧啶营养缺陷筛选标记URA3,柯巴基焦磷酸合酶和次丹参酮二烯合酶基因的启动子均为酿酒酵母的半乳糖激酶1启动子GAL1。
所述携带3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2基因的质粒,复制位点为2MICRON和CEN6/ARSH4中的一种,筛选标记为亮氨酸营养缺陷筛选标记LEU2和基因素(G418)抗性筛选标记KanMX中的一种,3-羟基-3-甲基戊二酰辅酶A还原酶基因的启动子为酿酒酵母的乙醇脱氢酶1启动子ADH1,萜类调控因子蛋白UPC基因的启动子为酿酒酵母的翻译延伸因子1启动子TEF1。
所述携带牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶基因的质粒,复制位点为2MICRON和CEN6/ARSH4中的一种,筛选标记为组氨酸营养缺陷筛选标记HIS3和潮霉素筛选标记hphNT1中的一种。一种牻牛儿牻牛儿基焦磷酸合酶基因和法呢基焦磷酸合酶基因融合在一起,启动子为酿酒酵母的乙醇脱氢酶1启动子ADH1,另一种牻牛儿牻牛儿基焦磷酸合酶基因的启动子为酿酒酵母的3-磷酸甘油酸激酶启动子PGK1。
7.根据权利要求2-6中任一所述的方法,其特征在于:
所述柯巴基焦磷酸合酶为丹参的柯巴基焦磷酸合酶。
所述次丹参酮二烯合酶为丹参的次丹参酮二烯合酶。
所述萜类调控因子蛋白UPC2为酿酒酵母固醇调控因子蛋白UPC2的突变。
所述3-羟基-3-甲基戊二酰辅酶A还原酶为酿酒酵母的3-羟基-3-甲基戊二酰辅酶A还原酶1截取5’部分序列的功能蛋白。
所述牻牛儿牻牛儿基焦磷酸合酶一种为酿酒酵母的牻牛儿牻牛儿基焦磷酸合酶;另一种为密码子优化的嗜酸热硫化叶菌的牻牛儿牻牛儿基焦磷酸合酶,其编码基因为序列表中序列1所示DNA。所述法呢基焦磷酸合酶为酿酒酵母的法呢基焦磷酸合酶。
所述出发酿酒酵母为酿酒酵母Saccharomyces cerevisiae BY4742。
8.由权利要求2-7中任一所述的方法构建得到的重组酿酒酵母。
9.权利要求1所述的酿酒酵母或权利要求8所述重组酿酒酵母在生产次丹参酮二烯中的应用。
10.一种生产次丹参酮二烯的方法,包括以下步骤:发酵权利要求1所述的酿酒酵母或权利要求8所述重组酿酒酵母,得到次丹参酮二烯。
11.根据权利要求10所述的方法,其特征在于:
所述发酵的温度为25℃-37℃或25℃或30℃或32℃或37℃;
所述发酵的体系的pH值为3.0-8.0或3.0或4.0或5.0或6.0或7.0或8.0;
所述发酵的时间为24-168小时或24小时或48小时或72小时或96小时或120小时或144小时或168小时;
所述发酵的接种量的体积百分比为0.01%-10%或0.01%或0.3%或1%或10%;
所述发酵的培养基由以下成分组成:
酵母膏、蛋白胨、葡萄糖、基因素(G418)、潮霉素
以上成分在所述发酵培养基中的浓度分别为:
酵母膏1-20g/L、蛋白胨1-40g/L、葡萄糖5-50g/L、G41810-500mg/L、潮霉素10-600mg/L。
说明书
技术领域
本发明涉及生产次丹参酮二烯的酿酒酵母基因工程菌及其构建方法与应用。
背景技术
次丹参酮二烯(Miltiradiene)是重要药用植物丹参中提取的脂溶性二萜成分,为丹参酮类化合物共同的前体物质(Wei Gao et al.,2009,Organic Letters,11:5170-5173)。这类化合物(如隐丹参酮、丹参酮IIA、丹参酮IIB、二氢丹参酮)在抗炎,心脑血管,抗肿瘤等方面有非常好的应用前景,目前与其相关的产品中,已有复方丹参片、复方丹参滴丸等销售额上数亿人民币的王牌产品上市。注射用丹参酮IIA磺酸钠已广泛用于治疗冠心病、心肌梗死等疾病,就目前丹参酮IIA的原料市场情况来看,含量20%的4500元/kg;含量50%的8000元/kg;含量为98%的高达3.6万元/kg。可见整个与丹参酮类化合物相关的产业有巨大的市场需求,经济效益也非常可观。目前丹参酮类化合物的主要来源是通过中药材丹参中直接提取,但随着开垦荒地、荒山伐林丹参野生资源的生长环境遭到严重破坏,丹参资源日趋减少;人工种植过程中也遇到品种退化,大量的土地和人力成本等因素。丹参酮类化合物产量已经远不能满足社会的需求,严重影响了丹参的临床应用和丹参酮类制药原料中间体的开发和应用,亟待需要提供新的资源途径。
目前利用合成生物学的原理,设计和改造微生物菌株来生产天然产物已被国际认为是一种最有潜力的方法,如在大肠杆菌中生产紫杉醇的前体紫杉二烯已达到1000mg/L(Parayil Kumaran Ajikumar et al.,2010,Science,330:70-74);银杏内酯类(Ginkgolides)前体左旋海松二烯(Levopimaradiene),在改造后的大肠杆菌工程菌中达到700mg/L的产量(EffendiLeonard et al.,2010,PNAS,107(31):13654-13659);在酵母工程菌中生产青蒿素(Artemisinin)的前体青蒿酸(Artemisinic acid)最高达到100mg/L(Dae-Kyun Ro et al.,2006,Nature,440:940-943);目前国内在青蒿素和紫杉醇等药物分子的生物合成方面有相关研究。
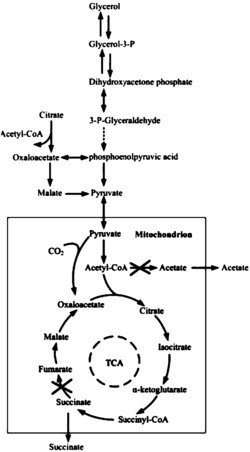
次丹参酮二烯为植物丹参体内萜类合成途径的产物,由丹参细胞中线粒体、细胞溶质和内质网存在的甲羟戊酸(MVA Pathway)代谢途径与在质体中存在的丙酮酸/磷酸甘油醛(MEP Pathway)代谢途径共同合成。其中前体物质牻牛儿基牻牛儿基焦磷酸(GGPP)为异戊二烯焦磷酸(IPP)和二甲基烯丙基焦磷酸(DMAPP)经过牻牛儿基牻牛儿基焦磷酸合酶(GGPS)催化得到,GGPP可以被丹参柯巴基焦磷酸合酶(SmCPS)和次丹参酮二烯合酶(SmKSL)共同催化为次丹参酮二烯,图1。微生物大肠杆菌中存在丙酮酸/磷酸甘油醛途径,在前面的研究中, 高伟等人研究策略采用了文献报道的大肠杆菌表达系统,以商用双表达载体pACYCDuet共同表达基因SmCPS和SmKSL,但效果不理想,只能获得了毫克级的次丹参酮二烯(Wei Gao et al.,2009,Organic Letters,11:5170-5173)。然而,作为传统发酵工艺中常用菌株:酿酒酵母,在其体内存在生产萜类成分的甲羟戊酸途径,其中产萜类成分麦角甾醇(Ergosterol)能达到生物量的4.6%(Arnezeder,C.et al.,1990,Biotechnol lett.,12:277-282);二萜香叶酰香叶酰醇(GGOH)也能达到283mg/L(Tokuhiro,K.et al.,2009,Appl Environ Microbiol.,75:5536-5543),由此利用酵母的优化的甲羟戊酸途径生产次丹参酮二烯具有很大潜力。
发明内容
本发明的一个目的是提供高产次丹参酮二烯酿酒酵母工程菌株ZD-Tans-001。
本发明所提供的酿酒酵母工程菌株ZD-Tans-001,其保藏号为CGMCC No.5296。
本发明的另一个目的是提供一种基因工程菌的构建方法。
本发明所提供的基因工程菌的构建方法,包括以下步骤:
在出发酿酒酵母中引入外源的柯巴基焦磷酸合酶和次丹参酮二烯合酶,得到重组酿酒酵母,记作重组酿酒酵母I。
所述方法还包括以下步骤:
在所述重组酿酒酵母I中提高3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2的活性,得到重组酿酒酵母,记作重组酿酒酵母II。
在所述重组酿酒酵母I中提高牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶的活性,得到重组酿酒酵母,记作重组酿酒酵母III。
所述方法还包括以下步骤:
在所述重组酿酒酵母III中提高3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2的活性,得到重组酿酒酵母,记作重组酿酒酵母IV。
所述方法还包括以下步骤:
克隆及人工改造萜类合成途径中功能基因:酿酒酵母3-羟基-3-甲基戊二酰辅酶A还原酶基因1(tHMG1)、酿酒酵母萜类调控因子蛋白UPC2基因(UPC2.1)、酿酒酵母牻牛儿牻牛儿基焦磷酸合酶基因(BTS1)和酿酒酵母法呢基焦磷酸合酶基因(ERG20);全人工合成嗜酸热硫化叶菌的牻牛儿牻牛儿基焦磷酸合酶基因(SaGGPSsyn)。克隆营养缺陷型筛选标记:亮氨酸营养缺陷筛选基因元件LEU2、组氨酸营养缺陷筛选基因元件HIS3、尿嘧啶营养缺陷筛选基因元件URA3;抗生素型筛选标记基因素(G418)抗性基因元件KanMX和潮霉素抗性基因元件hphNT1等功能元件。
所述方法还包括以下步骤:
克隆调控元件酿酒酵母的3-磷酸甘油酸激酶启动子(PGK1)、酿酒酵母的翻译延伸因子1启动子(TEF1)、酿酒酵母的乙醇脱氢酶1启动子(ADH1)、酿酒酵母的半乳糖激酶1启动子(GAL1)、酿酒酵母细胞色素C终止子(CYC1t)和酿酒酵母的乙醇脱氢酶1终止子(ADH1t)。
所述引入外源的柯巴基焦磷酸合酶和次丹参酮二烯合酶是通过转化入携带外源柯巴基焦磷酸合酶和次丹参酮二烯合酶基因的质粒,或在酿酒酵母染色体上整合外源柯巴基焦磷酸合酶和次丹参酮二烯合酶基因。
所述提高3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2的活性是通过转化入携带3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2基因的质粒,或在酿酒酵母染色体上整合3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2基因。
所述提高牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶的活性是通过转化入携带牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶基因的质粒,或在酿酒酵母染色体上整合牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶基因。
所述在酿酒酵母染色体上整合基因是整合在δDNA位点。
所述携带柯巴基焦磷酸合酶和次丹参酮二烯合酶基因的质粒,复制位点为2MICRON,筛选标记为尿嘧啶营养缺陷筛选标记URA3,柯巴基焦磷酸合酶和次丹参酮二烯合酶基因的启动子均为酿酒酵母的半乳糖激酶1启动子GAL1。
所述携带3-羟基-3-甲基戊二酰辅酶A还原酶和萜类调控因子蛋白UPC2基因的质粒,复制位点为2MICRON和CEN6/ARSH4中的一种,筛选标记为亮氨酸营养缺陷筛选标记LEU2和基因素(G418)抗性筛选标记KanMX中的一种,3-羟基-3-甲基戊二酰辅酶A还原酶基因的启动子为酿酒酵母的乙醇脱氢酶1启动子ADH1,萜类调控因子蛋白UPC基因的启动子为酿酒酵母的翻译延伸因子1启动子TEF1。
所述携带牻牛儿牻牛儿基焦磷酸合酶和法呢基焦磷酸合酶基因的质粒,复制位点为2MICRON和CEN6/ARSH4中的一种,筛选标记为组氨酸营养缺陷筛选标记HIS3和潮霉素筛选标记hphNT1中的一种。一种牻牛儿牻牛儿基焦磷酸合酶基因和法呢基焦磷酸合酶基因融合在一起,启动子为酿酒酵母的乙醇脱氢酶1启动子ADH1,另一种牻牛儿牻牛儿基焦磷酸合酶基因的启动子为酿酒酵母的3-磷酸甘油酸激酶启动子PGK1。
所述柯巴基焦磷酸合酶为丹参的柯巴基焦磷酸合酶。
所述次丹参酮二烯合酶为丹参的次丹参酮二烯合酶。
所述萜类调控因子蛋白UPC2为酿酒酵母固醇调控因子蛋白UPC2的突变。
所述3-羟基-3-甲基戊二酰辅酶A还原酶为酿酒酵母的3-羟基-3-甲基戊二酰辅酶A还原酶1截取5’部分序列的功能蛋白。
所述牻牛儿牻牛儿基焦磷酸合酶一种为酿酒酵母的牻牛儿牻牛儿基焦磷酸合酶;另一种为密码子优化的嗜酸热硫化叶菌的牻牛儿牻牛儿基焦磷酸合酶,其编码基因为序列表中序列1所示DNA。所述法呢基焦磷酸合酶为酿酒酵母的法呢基焦磷酸合酶。
所述出发酿酒酵母为酿酒酵母Saccharomyces cerevisiae BY4742。
由所述方法构建得到的重组酿酒酵母也属于本发明的保护范围。
所述酿酒酵母菌ZD-Tans-001或所述酿酒酵母菌在生产次丹参酮二烯中的应用也属于本发明的保护范围。
本发明的又一个目的是提供一种生产次丹参酮二烯的方法。
本发明所提供的生产次丹参酮二烯的方法,包括以下步骤:
发酵所述酿酒酵母ZD-Tans-001或所述酿酒酵母,得到次丹参酮二烯。
所述发酵的温度为25℃-37℃或25℃或30℃或32℃或37℃;
所述发酵的体系的pH值为3.0-8.0或3.0或4.0或5.0或6.0或7.0或8.0;
所述发酵的时间为24-168小时或24小时或48小时或72小时或96小时或120小时或144小时或168小时;
所述发酵的接种量的体积百分比为0.01%-10%或0.01%或0.3%或1%或10%;
所述发酵的培养基由以下成分组成:
酵母膏、蛋白胨、葡萄糖、基因素(G418)、潮霉素
以上成分在所述发酵培养基中的浓度分别为:
酵母膏1-20g/L、蛋白胨1-40g/L、葡萄糖5-50g/L、G41810-500mg/L、潮霉素10-600mg/L。
本发明所构建的酿酒酵母ZD-Tans-001在好养氧条件下,发酵6天时可生产487.91mg/L的次丹参酮二烯。
保藏说明
菌种名称:酿酒酵母
拉丁名:Saccharomyces cerevisiae
菌株编号:ZD-Tans-001
保藏机构:中国微生物菌种保藏管理委员会普通微生物中心
保藏机构简称:CGMCC
地址:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所
保藏日期:2011年9月28日
保藏中心登记入册编号:CGMCC No.5296
附图说明
图1次丹参酮二烯生物合成途径
图2携带基因元件的质粒示意图(营养缺陷型筛选标记)
图3携带基因元件的质粒示意图(抗生素型筛选标记)
图4次丹参酮二烯GC-MS分析图A:次丹参酮二烯标准品TIC图(Rt=19.922min);B:样品TIC图(Rt=19.919min);C:次丹参酮二烯标准品MS图;D:次丹参酮二烯 样品MS图
图5菌株发酵结果:与ZD-T-000相关菌株
图6菌株发酵结果:与ZD-T-010及转入营养缺陷筛选标记型功能质粒相关菌株
图7菌株发酵结果:与ZD-T-010及转入抗生素筛选标记型功能质粒相关菌株
图8ZD-Tans-001菌株高密度发酵结果
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1、基因元件的克隆
基因元件的克隆分为以下五个步骤:
(1)酵母DNA提取
挑取菌斑(Saccharomyces cerevisiae BY4742)于YPD液体培养基(配方:1%Yeast Extract(酵母膏),2%Peptone(蛋白胨),2%Dextrose(葡萄糖)中,30℃,200rpm,培养24h。10000g,5分钟收集菌体于1.5ml离心管中,水清洗两次,菌体重悬于酵母破壁液中(25ul酵母破壁酶,470ul山梨醇缓冲液,5ul β-ME),30℃温浴1h后离心;菌体用500ul TENTS缓冲液(10mM Tris-HCl,pH 7.5;1mM EDTA,pH8.0;100mMNaAc;2%triton-100;1%SDS)重悬,60℃水浴1h;酚/氯仿抽提2次;上清液加3倍体积的EtOH,1/10倍体积的3M NaAc,-20℃冰箱放置2h;13000g,4℃,离心10min,倒掉上清,沉淀用70%EtOH,洗剂沉淀2次后吹干,双蒸水溶解,-20℃保存备用。
(2)PCR扩增及克隆
以酵母基因组DNA为模板,用引物列表1中引物,扩增tHMG1、UPC2、δDNA1、δDNA1;以质粒YES2.0DNA为模板扩增营养缺陷型筛选标记URA3;以质粒pRS313DNA为模板扩增营养缺陷型筛选标记HIS3;以质粒pRS425DNA为模板扩增营养缺陷型筛选标记LEU2;以质粒pRS41KDNA为模板扩增抗生素型筛选标记KanMX;以质粒pRS42HDNA为模板扩增抗生素型筛选标记hphNT1。扩增体系为:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、引物(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。
引物列表1
扩增条件为98℃预变性2分钟(1个循环);98℃变性10秒、退火10秒(退火温度见引物列表1)、72℃延伸均用2分钟(32个循环);72℃延伸8分钟(1个循环)。将扩增产物克隆到pEASY-Blunt克隆载体(购自北京全式金生物技术有限公司)上。克隆体系为:1ul PCR扩增产物、1ul pEASY-Blunt克隆载体,轻轻混合、室温反应10分钟后加入50ul Trans10感受态细胞中(购自北京全式金生物技术有限公司),冰浴30分钟。42℃热激30秒,立即置于冰上2分钟。加入250ul LB培养基,100rpm,37℃孵育1小时。取200ul菌液涂在含有氨苄青霉素的LB平板上,过夜培养后,PCR筛选5个阳性单菌落,将阳性克隆进行液体培养,提取阳性克隆质粒进行测序验证,测序结果表明在载体pEASY-Blunt上插入目的片段。
(3)UPC2基因突变
UPC2是能调控酵母体内萜类水平的一个关键转录因子基因,UPC2基因3’端(2669bp)的G到A突变,可以使相应位点的氨基酸从Glycine变为Aspartate,在有氧条件下能够提高细胞内萜类的含量,我们用基因Quickchange的点突变方法将此位点突变:首先在含盖突变位点区设计同源互补且将G改为A的引物UPC(G-A)-F和UPC(G-A)-R(引物列表2),利用第二步中得到的pEASY-Blunt-UPC2质粒为模板,扩增体系为:10×PfuUltra II reaction buffer 5ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、引物(10uM)各1ul、Stratagene PfuUltra II fusion HS DNA polymerase 1ul、补加蒸馏水至总体积50ul。扩增条件为95℃预变性2分钟(1个循环);95℃变性30秒、退火30秒(退火温度见引物列表1)、72℃延伸均用6分钟(32个循环);72℃延伸8分钟(1个循环)。产物纯化后,用DpnI酶处理后,转入大肠感受态Trans10,涂板后经过抗性和PCR验证,测序鉴定成功突变的基因,获得突变基因UPC2.1。
引物列表2
(4)BTS1和ERG20基因的融合
酿酒酵母BTS1基因表达的牻牛儿牻牛儿基焦磷酸合酶能催化酿酒酵母ERG20基因表达的法呢基焦磷酸合酶的产物法呢基焦磷酸(FPP)为牻牛儿牻牛儿基焦磷酸(GGPP)。两酶在物理上的融合能够增加FPP转化为GGPP的催化活性,我们用GGGS肽将BTS1和ERG20两个功能蛋白融合,以提高其催化效率。以第一步中提取的DNA为模板,用引物列表3中引物,扩增体系为:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、引物(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性2分钟(1个循环);98℃变性10秒、退火10秒(退火温度见引物列表3)、72℃延伸均用2分钟(32个循环);72℃延伸8分钟(1个循环)。割胶纯化两个目的片段,各50ng加入PCR体系:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、加引物Asc1-FPS和SexA-Bst/DPP(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性2分钟(1个循环);98℃变性10秒、58℃退火10秒、72℃延伸均用 2分钟(32个循环);72℃延伸8分钟(1个循环);将目标大小片段扩增产物克隆到pEASY-Blunt克隆载体。转化,测序验证(方法同步骤2)。
引物列表3
(5)SaGGPSSyn.基因密码子优化与基因全合成
嗜酸热硫化叶菌(Sulfolobus acidocaldarius)的牻牛儿牻牛儿基焦磷酸合酶能直接催化IPP和DMAPP转化为GGPP(通常的牻牛儿牻牛儿基焦磷酸合酶,如酿酒酵母,催化FPP和IPP合成GGPP)。我们将其基因优化为酵母适合的密码子、GC含量为38.93%、5’端加酶切位点SexA1,3’端加酶切位点Asc1后基因全合成基因序列1。保存在载体(pUC57)中,得到SaGGPSSyn.基因。
实施例2、调控元件的克隆
以酿酒酵母基因组DNA为模板,用引物列表4中引物,扩增PGK1(750bp)、TEF1(450bp)和ADH1(1500bp)启动子,及终止子ADH1t(158bp)。扩增体系为:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、引物(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性2分钟(1个循环);98℃变性10秒、退火10秒(退火温度见引物列表4)、72℃延伸1.5分钟(32个循环);72℃延伸8分钟(1个循环)。将扩增产物克隆到pEASY-Blunt克隆载体上。转化,测序验证(方法同实施例1步骤2)。
引物列表4
实施例3、携带基因元件的质粒构建
携带基因元件的质粒构建分为以下两个步骤
(1)携带tHMG1-UPC2.1基因的质粒构建
携带tHMG1-UPC2.1基因的质粒构建,共有以下8步:
第一步:pRS406-δDNA-URA3质粒的构建。
用BstE1和Sac1酶切实施例1步骤2中获得的质粒pEASY-Blunt-δDNA1;BstE1和Kpn1酶切实施例1步骤2中获得的质粒pEASY-Blunt-δDNA2。割胶纯化两个目的片段,各50ng加入连接体系:2ul 10XT4ligation Buffer(NEB公司)、1ul T4ligase(NEB公司,400,000cohesive end units/ml),补充蒸馏水至20ul,室温反应2小时得到连接产物,取1ul连接产物加入PCR体系:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、加引物Sac1-A-δDNA1和Kpn-A-δDNA2(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性1.5分钟(1个循环);98℃变性10秒、58℃退火10秒、72℃延伸均用1分钟(32个循环);72℃延伸8分钟(1个循环);将目标大小片段扩增产物克隆到pEASY-Blunt克隆载体,转化,测序验证(方法同实施例1步骤2)。
Sac1和Kpn1双酶切质粒pEASY-Blunt-δDNA和pRS406,割胶回收目的片段:δDNA(100ng)和pRS406(30ng),加2ul 10XT4ligation Buffer(NEB公司)、1ul T4 ligase(NEB公司,400,000cohesive end units/ml),补充蒸馏水至20ul,室温反应2小时得到连接产物。转入Trans10感受态细胞,测序验证(方法同实施例1步骤2),得到pRS406-δDNA质粒。
BstE1酶切质粒pRS406-δDNA和pEASY-Blunt-URA3,割胶回收目的片段:URA3(100ng)和pRS406-δDNA(50ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到pRS406-δDNA-URA3质粒。
第二步:pRS406-δDNA-URA3-ADH1-tHMG1-ADH1t质粒的构建
用SexA1和Sac11酶切实施例2中获得的质粒pEASY-Blunt-ADH1;SexA1和Asc1酶切实施例1步骤2中获得的质粒pEASY-Blunt-tHMG1,割胶纯化两个目的片段,各50ng加入 连接体系:2ul 10XT4ligation Buffer(NEB公司)、1ul T4ligase(NEB公司,400,000cohesive end units/ml),补充蒸馏水至20ul,室温反应2小时得到连接产物,取1ul连接产物加入PCR体系:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、加引物Sac11-Pac1-ADH-2和Asc1-HMG1(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性2分钟(1个循环);98℃变性10秒、58℃退火10秒、72℃延伸均用1.5分钟(32个循环);72℃延伸8分钟(1个循环);将目标大小片段扩增产物克隆到pEASY-Blunt克隆载体,转化,测序验证(方法同实施例1步骤2)。得到pEASY-Blunt-ADH1-tHMG1质粒。
Sac11和Asc1双酶切质粒pEASY-Blunt-ADH1-tHMG1;Asc1和Pme1酶切实施例2中质粒pEASY-Blunt-ADH1t;割胶纯化两个目的片段,各50ng加入连接体系,连接体系同上,室温反应2小时得到连接产物,取1ul连接产物加入PCR体系:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、加引物Sac11-Pac1-ADH-2和Sac11-Pme-ADHt(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性2分钟(1个循环);98℃变性10秒、58℃退火10秒、72℃延伸均用2.5分钟(32个循环);72℃延伸8分钟(1个循环);将目标大小片段扩增产物克隆到pEASY-Blunt克隆载体,转化,测序验证(方法同实施例1步骤2)。得到pEASY-Blunt-ADH1-tHMG1-ADH1t质粒。
Sac11分别酶切质粒pEASY-Blunt-ADH1-tHMG1-ADH1t和pRS406-δDNA-URA3,割胶回收目的片段:ADH1-tHMG1-ADH1t(100ng)和pRS406-δDNA-URA3(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到pRS406-δDNA-URA3-ADH1-tHMG1-ADH1t质粒。
第三步:pHUra-δDNA-tHMG1-UPC2.1质粒的构建
SexA1和Pac1双酶切质粒pRS406-δDNA-URA3-ADH1-tHMG1-ADH1t和pEASY-Blunt-TEF1,割胶回收目的片段:pRS406-δDNA-URA3-//-tHMG1-ADH1t(100ng)和TEF1(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pRS406-δDNA-URA3-TEF1-tHMG1-ADH1t。
SexA1和Asc1双酶切质粒pRS406-δDNA-URA3-TEF1-tHMG1-ADH1t和pEASY-Blunt-UPC2.1,割胶回收目的片段:pRS406-δDNA-URA3-TEF1-//-ADH1t(100ng)和UPC2.1(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pRS406-δDNA-URA3-TEF1-UPC2.1-ADH1t。
Sac11-Pme-ADHt和pme-pTEF1为引物(引物列表4,5),质粒pRS406 -δDNA-URA3-TEF1-UPC2.1-ADH1t为模板,PCR获得DNA片段:TEF1-UPC2.1-ADH1t。
Pme1酶切质粒pRS406-δDNA-URA3-ADH1-tHMG1-ADH1t和DNA片段TEF1-UPC2.1-ADH1t,割胶回收目的片段:pRS406-δDNA-URA3-ADH1-tHMG1-ADH1t(100ng)和TEF1-UPC2.1-ADH1t(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHUra-δDNA-tHMG1-UPC2.1。
第四步:pLHis-tHMG1-UPC2.1质粒的构建
Sac11酶切质粒pHUra-δDNA-tHMG1-UPC2.1和pRS313,割胶回收目的片段:ADH1-tHMG1-ADH1t-TEF1-UPC2.1-ADH1t(100ng)与pRS313(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pLHis-tHMG1-UPC2.1。
第五步:pHLeu-tHMG1-UPC2.1质粒的构建
Sac11酶切质粒pHUra-δDNA-tHMG1-UPC2.1和pRS425,割胶回收目的片段:ADH1-tHMG1-ADH1t-TEF1-UPC2.1-ADH1t(100ng)与pRS425(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHLeu-tHMG1-UPC2.1。
第六步:pLKanMX-tHMG1-UPC2.1质粒的构建
取实施例1获得的基因元件KanMX,取30ng纯化后的PCR扩增产物,加入2ul 10XT4ligation Buffer(NEB公司)、1ul T4Polynucleotide kinase(NEB公司),补充蒸馏水至20ul,37℃磷酸化60分钟;同以质粒pLHis-tHMG1-UPC2.1为模板,V313-to-R和V313-to-F为引物PCR获得去Maker反向部分(100ng),T4连接酶连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pLKanMX-tHMG1-UPC2.1。
第七步:pHKanMX-tHMG1-UPC2.1质粒的构建
取实施例1获得的基因元件KanMX,取30ng纯化后的PCR扩增产物,加入2ul 10XT4ligation Buffer(NEB公司)、1ul T4Polynucleotide kinase(NEB公司),补充蒸馏水至20ul,37℃磷酸化60分钟;同以质粒pHLeu-tHMG1-UPC2.1为模板,V-425-to-R和V-425-to-F为引物PCR获得pRS425去Maker反向部分(100ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHKanMX-tHMG1-UPC2.1。
第八步:pLLeu-tHMG1-UPC2.1质粒的构建
取实施例1获得的基因元件LEU2元件,取30ng纯化后的PCR扩增产物,加入2ul10XT4ligation Buffer(NEB公司)、1ul T4Polynucleotide kinase(NEB公司),补充蒸馏水至20ul,37℃磷酸化60分钟;同以质粒pLHis-tHMG1-UPC2.1为模板,V313-to-R和V313-to-F为引物PCR获得去Maker反向部分(100ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pLLeu-tHMG1-UPC2.1。
(2)携带ERG20、BTS1和SaGGPS Syn.基因的质粒构建
携带ERG20、BTS1和SaGGPS Syn.基因的质粒构建,共有以下7步:
第一步:pHUra-δDNA-ERG20/BTS1-SaGGPSSyn.质粒的构建
SexA1和Asc1双酶切pRS406-δDNA-URA3-ADH1-tHMG1-ADH1t和质粒pEASY-Blunt-ERG20/BTS1,割胶回收目的片段:pRS406-δDNA-URA3-ADH1-//-ADH1t(100ng)和ERG20/BTS1(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pRS406-δDNA-URA3-ADH1-ERG20/BTS1-ADH1t。
SexA1和Pac1双酶切pRS406-δDNA-URA3-ADH1-tHMG1-ADH1t质粒和pEASY-Blunt-PGK1,割胶回收目的片段:pRS406-δDNA-URA3-//-tHMG1-ADH1t(100ng)和PGK1(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pRS406-δDNA-URA3-PGK1-tHMG1-ADH1t。
SexA1和Asc1双酶切pRS406-δDNA-URA3-PGK1-tHMG1-ADH1t和实施例1步骤5获得的质粒pUC57-SaGGPSSyn.,割胶回收目的片段:
pRS406-δDNA-URA3-PGK1-//-ADH1t(100ng)和SaGGPSSyn.(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pRS406-δDNA-URA3-PGK1-SaGGPSSyn.-ADH1t。Sac11-Pme-ADHt和pme-pPGK1为引物,质粒pRS406-δDNA-URA3-PGK1-SaGGPSSyn.-ADH1t为模板PCR获得:PGK1-SaGGPSSyn.-ADH1t。
Pme1酶切质粒pRS406-δDNA-URA3-ADH1-ERG20/BTS1-ADH1t和PGK1-SaGGPSSyn.-ADH1t,割胶回收目的片段:pRS406-δDNA-URA3-ADH1-ERG20/BTS1-ADH1t(100ng)和PGK1-SaGGPSSyn.-ADH1t(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHUra-δDNA-ERG20/BTS1-SaGGPSSyn.。
第二步:pLUra-ERG20/BTS1-SaGGPSSyn.质粒的构建
Sac11酶切质粒pHUra-δDNA-ERG20/BTS1-SaGGPSSyn.和pRS316,割胶回收目的片段:ADH1-ERG20/BTS1-ADH1t-PGK1-SaGGPSSyn.-ADH1t(100ng)与pRS316(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:
pLUra-ERG20/BTS1-SaGGPSSyn.。
第三步:pHUra-ERG20/BTS1-SaGGPSSyn.质粒的构建
Sac11酶切质粒pHUra-δDNA-ERG20/BTS1-SaGGPSSyn.和pRS426,割胶回收目的片段:ADH1-ERG20/BTS1-ADH1t-PGK1-SaGGPSSyn.-ADH1t(100ng)与pRS426(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHUra-ERG20/BTS1-SaGGPSSyn.。
第四步:pLhphNT1-ERG20/BTS1-SaGGPSSyn.质粒的构建
取实施例1获得的基因元件hphNT1,取30ng纯化后的PCR扩增产物,加入2ul10XT4ligation Buffer(NEB公司)、1ul T4Polynucleotide kinase(NEB公司),补充蒸馏水至20ul,37℃磷酸化60分钟;同以质粒pLUra-ERG20/BTS1-SaGGPSSyn.为模板,V316-to-R和V316-to-F为引物PCR获得去Maker反向部分(100ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pLhphNT1-ERG20/BTS1-SaGGPSSyn.。
第五步:pHhphNT1-ERG20/BTS1-SaGGPSSyn.质粒的构建
取实施例1获得的基因元件hphNT1,取30ng纯化后的PCR扩增产物,加入2ul10XT4ligation Buffer(NEB公司)、1ul T4Polynucleotide kinase(NEB公司),补充蒸馏水至20ul,37℃磷酸化60分钟;同以质粒pHUra-ERG20/BTS1-SaGGPSSyn.为模板,V316-to-R和V316-to-F为引物PCR获得去Maker反向部分(100ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHhphNT1-ERG20/BTS1-SaGGPSSyn.。
第六步:pLHis-ERG20/BTS1-SaGGPSSyn.质粒的构建
取实施例1获得的基因元件HIS3,取30ng纯化后的PCR扩增产物,加入2ul 10XT4ligation Buffer(NEB公司)、1ul T4Polynucleotide kinase(NEB公司),补充蒸馏水至20ul,37℃磷酸化60分钟;同以质粒pLhphNT1-ERG20/BTS1-SaGGPSSyn.为模板,V316-to-R和V316-to-F为引物PCR获得去Maker反向部分(100ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pLHis-ERG20/BTS1-SaGGPSSyn.。
第七步:pHHis-ERG20/BTS1-SaGGPSSyn.质粒的构建
取实施例1获得的基因元件HIS3,取30ng纯化后的PCR扩增产物,加入2ul 10XT4ligation Buffer(NEB公司)、1ul T4Polynucleotide kinase(NEB公司),补充蒸馏水至20ul,37℃磷酸化60分钟;同以质粒pHhphNT1-ERG20/BTS1-SaGGPSSyn.为模板,V316-to-R和V316-to-F为引物PCR获得去Maker反向部分(100ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHHis-ERG20/BTS1-SaGGPSSyn.。
引物表5
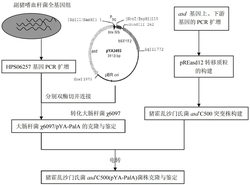
实施例4、酿酒酵母基因工程菌ZD-T-000的构建
酿酒酵母基因工程菌ZD-T-000的构建分为以下两个步骤:
(1)携带SmCPS和SmKSL基因质粒的构建
质粒模板pUC19-SmCPS,引物Kpn-CPS-2和CPS-Not,扩增SmCPS;质粒模板pUC19-SmKSL,引物2-pvul-KSL和KSL-Pme1,扩增SmKSL;质粒模板YES2/CT,引物Kpn-GAL1和2-GAL1-pvul,扩增GAL1;引物Not1-CYCt和CYCt-Kpn1扩增CYC1t,见引物列表6。扩增体系为:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、引物(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性1.5分钟(1个循环);98℃变性10秒、退火10秒(退火温度见引物列表6)、72℃延伸均用2分钟(32个循环);72℃延伸8分钟(1个循环),产物经割胶回收保存。
用Kpn 1和Not 1双酶切质粒YES2/CT和片段SmCPS,割胶回收目的片段:YES2/CT(100ng)和SmCPS(30ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:YES2/CT-GAL1-SmCPS-CYC1t
用Kpn 1和pvul酶切基因片段SmKSL、GAL1、CYC1t,分别割胶回收目的片段后,SmKSL(100ng)、GAL1(30ng)和CYC1t(30ng)混合加入连接体系,连接体系同上,室温反应2小时得到连接产物,取1ul连接产物加入PCR体系:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、加引物Not1-CYCt和KSL-Pme1(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性2分钟(1个循环);98℃变性10秒、58℃退火10秒、72℃延伸均用2分钟(32个循环);72℃延伸8分钟(1个循环);将目标大小片段扩增产物克隆到pEASY-Blunt克隆载体,pEASY-Blunt-CYC1t-GAL1-SmKSL,转化,测序验证(方法同实施例1步骤2)。
Not1和Pme1双酶切质粒YES2/CT-GAL1-SmCPS-CYC1t和pEASY-Blunt-CYC1t-GAL1-SmKSL,割胶回收目的片段YES2/CT-GAL1-SmCPS-CYC1t(50ng)和CYC1t-GAL1-SmKSL(100ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHUra-SmCPS-SmKSL。
引物列表6
(2)携带SmCPS和SmKSL基因的质粒转化入酿酒酵母
出发菌酿酒酵母Saccharomyces cerevisiae BY4742于YPD中过夜培养,取1ml(0D约0.6-1.0)分装到1.5ml EP管中,4℃、10000g离心1min,弃上清,沉淀用无菌水(4℃)洗涤,同样条件下离心,弃上清。菌体加入1ml处理液(10mM LiAc;10mM DTT;0.6M sorbitol;10mM Tris-HCl(pH7.5),处理液使用时才加DTT),25℃下放置20min。离心,弃上清,菌体中加入1ml 1M sorbitol(0.22um水系膜过膜除菌)重悬,离心,弃上清(用1M sorbitol重悬二次),到最终体积约为90μl。加入5ug转化用质粒:pHUra-SmCPS-SmKSL混匀后转入电击杯(0.2cm)中,冰浴5min.,3ky,25μF,200Ω,电击一次。电击后立刻加入1mL 1M sorbitol,吸出后于30℃培养1h。菌液经离心处理后全部涂在选择平板上,配方:0.8%酵母选择培养基SD-Ura-Trp-His(北京泛基诺(功能基因组)科技有限公司),2%葡萄糖,0.005%His.,0.01%Trp.。30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-000
实施例5、酿酒酵母基因工程菌ZD-T-001,ZD-T-002,ZD-T-003和ZD-T-004的构建
采用和实施例4中相同的方法进行ZD-T-000感受态细胞的制备及转化,转入质粒pLLeu-tHMG1-UPC2.1。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.005%His.,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-001。
采用和实施例4中相同的方法进行ZD-T-000感受态细胞的制备及转化,转入质粒pHLeu-tHMG1-UPC2.1。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.005%His.,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-002。
采用和实施例4中相同的方法进行ZD-T-000感受态细胞的制备及转化,转入质粒pLHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-003。
采用和实施例4中相同的方法进行ZD-T-000感受态细胞的制备及转化,分别转入质粒pHHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-004。
实施例6、酿酒酵母基因工程菌ZD-T-005,ZD-T-006,ZD-T-007和ZD-T-008的构建
采用和实施例4中相同的方法进行ZD-T-000感受态细胞的制备及转化,转入质粒pLLeu-tHMG1-UPC2.1和pLHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-005。
采用和实施例4中相同的方法进行ZD-T-000感受态细胞的制备及转化,转入质粒pLLeu-tHMG1-UPC2.1和pHHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-006。
采用和实施例4中相同的方法进行ZD-T-000感受态细胞的制备及转化,转入质粒pHLeu-tHMG1-UPC2.1和pLHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-007。
采用和实施例4中相同的方法进行ZD-T-000感受态细胞的制备及转化,转入质粒pHLeu-tHMG1-UPC2.1和pHHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-008。
实施例7、用酿酒酵母基因工程菌ZD-T-000,ZD-T-001,ZD-T-002,ZD-T-003,ZD-T-004,ZD-T-005,ZD-T-007,ZD-T-007和ZD-T-008生产次丹参酮二烯
挑取在固体筛选培养平板中活化的工程菌株,于液体培养基中培养(30℃,250rpm,16小时),制备发酵种子液。离心收集菌体,转移至20ml发酵三角瓶中,调OD至0.1,各菌株相应的发酵培养基同筛选培养基(2%葡萄糖替换为2%半乳糖),30℃,250rpm/min.,振荡培养4天,检查OD600,及次丹参酮二烯的含量。
发酵液8000g收集菌体,加少量石英砂,500ul乙腈,Votex30秒,冰水中超声30min,7000g离心5min,取上清(提取三次,合并上清液);上清液过0.22um有机膜,GC-MS测定:进样口温度300℃,进样体积1ul,不分流,溶剂延时5min.;色谱柱:HP-5ms(30m*0.25*0.5um);色谱条件:50℃,2min;20℃min-1到250℃保温8min;MS条件:SIM:148、272;Rt:~12.90min,标准曲线定量分析,见图4。用高拷贝数质粒并诱导表达增加SmCPS-SmKSL基因表达的情况下(质粒pHUra-SmCPS-SmKSL),高拷贝的基因模块tHMG1,UPC2.1(pHLeu-tHMG1-UPC2.1);低拷贝的基因模块BTS1/ERG20,SaGGPSsyn.(pLHis-ERG20/BTS1-SaGGPSSyn.)得到的工程菌株ZD-T-007,产量为参照菌株ZD-T-000的12.5倍,达14.4mg/L,见图5。
实施例8、酿酒酵母基因工程菌ZD-T-010的构建
酿酒酵母基因工程菌ZD-T-010的构建分为以下两个步骤
(1):δDNA-URA3-TEF1-SmCPS-ADH1t-PGK1-SmKSL-ADH1t功能模块的构建
以质粒模板pUC19-SmCPS,SexA-CPS/KS和ASC1-CPS为引物,扩增SmCPS;质粒模板pUC19-SmKSL,SexA1-KSL和Asc1-CPS/KS为引物,扩增SmKSL。扩增体系为:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、引物(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性2分钟(1个循环);98℃变性10秒、退火10秒(退火温度见引物列表7)、72℃延伸均用2分钟(32个循环);72℃延伸8分钟(1个循环),产物清洗后用SexA1和Asc1双酶切,并割胶回收。
SexA1和Asc1双酶切质粒pRS406-δDNA-URA3-TEF1-UPC2.1-ADH1t和 pRS406-δDNA-URA3-PGK1-tHMG1-ADH 1t,并与相应的酶切片段SmCPS和SmKSL连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pRS406-δDNA-URA3-TEF1-SmCPS-ADH1t和pRS406-δDNA-URA3-PGK1-SmKSL-ADH1t
Sac11-Pme-ADHt和pme-pPGK1为引物,质粒pRS406-δDNA-URA3-PGK1-SmKSL-ADH1t为模板PCR获得,基因元件:PGK1-SmKSL-ADH1t。
Pme1酶切质粒pRS406-δDNA-URA3-TEF1-SmCPS-ADH1t和PGK1-SmKSL-ADH1t。割胶回收目的片段:pRS406-δDNA-URA3-TEF1-SmCPS-ADH1t(50ng)和PGK1-SmKSL-ADH1t(100ng),连接,转化,测序验证(方法同实施例3步骤1第一步)。得到质粒:pHUra-δDNA-SmCPS-SmKSL。
以质粒模板pHUra-δDNA-SmCPS-SmKSL,δDNA-F和δDNA-R-2为引物,扩增体系为:NewEngland Biolabs Phusion 5Xbuffer 10ul、dNTP(10mM each dNTP)1ul、DNA模板20ng、引物(10uM)各1ul、Phusion High-Fidelity DNA Polymerase(2.5U/ul)0.5ul、补加蒸馏水至总体积50ul。扩增条件为98℃预变性3钟(1个循环);98℃变性10秒、退火10秒(退火温度见引物列表7)、72℃延伸均用2分钟(32个循环);72℃延伸8分钟(1个循环),产物割胶回收备用。
引物列表7
(2)δDNA-URA3-TEF1-SmCPS-ADH1t-PGK1-SmKSL-ADH1t功能模块整合入酿酒酵母染色体
功能模块δDNA-URA3-TEF1-SmCPS-ADH1t-PGK1-SmKSL-ADH1t电击转入出发菌酿酒酵母Saccharomyces cerevisiae BY4742中,转化和筛选方法同实施例4步骤2。
PCR筛选出阳性克隆菌株,随机选取10株,发酵(碳源为2%葡萄糖),及产物检测同实施例7。选取次丹参酮二烯产量最高的菌株,命名为ZD-T-010。
实施例9、酿酒酵母基因工程菌ZD-T-011、ZD-T-012、ZD-T-013、ZD-T-014的构建(营 养缺陷型)
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pLLeu-tHMG1-UPC2.1。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.005%His.,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-011。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pHLeu-tHMG1-UPC2.1。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.005%His.,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-012。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pLHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-013。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,分别转入质粒pHHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-014。
实施例10、酿酒酵母基因工程菌ZD-T-015、ZD-T-016、ZD-T-017、ZD-T-018的构建(营养缺陷型)
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pLLeu-tHMG1-UPC2.1和pLHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-015。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pLLeu-tHMG1-UPC2.1和pHHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-016。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pHLeu-tHMG1-UPC2.1和pLHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-017。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pHLeu-tHMG1-UPC2.1和pHHis-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:0.8%酵母选择培养基SD-Ura-Trp-Leu-His,2%葡萄糖,0.01%Trp.;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-018。
实施例11、用酿酒酵母基因工程菌ZD-T-010,ZD-T-011,ZD-T-012,ZD-T-013,ZD-T-014,ZD-T-015,ZD-T-016,ZD-T-017和ZD-T-018生产次丹参酮二烯(营养缺陷型)
挑取在固体筛选培养平板中活化的工程菌株,于液体培养基中培养(30℃,250rpm,16小时),制备发酵种子液。离心收集菌体,转移至20ml发酵三角瓶中,调OD至0.1,各菌株相应的发酵培养基同筛选培养基,30℃,250rpm/min.,振荡培养4天,检查OD600,及次丹参酮二烯的含量。
发酵液8000g收集菌体,加少量石英砂,500ul乙腈,votex30秒,冰水中超声30min,7000g离心5min,取上清(提取三次,合并上清液);上清液过0.22um有机膜,GC-MS测定:进样口温度300℃,进样体积1ul,不分流,溶剂延时5min.;色谱柱:HP-5ms(30m*0.25*0.5um);色谱条件:50℃,2min;20℃min-1到250℃保温8min;MS条件:SIM:148、272;Rt:~12.90min,标准曲线定量分析。
在酿酒酵母Saccharomyces cerevisiae BY4742中整合δDNA-URA3-TEF1-SmCPS-ADH1t-PGK1-SmKSL-ADH1t基因的情况下,高拷贝的基因模块tHMG1,UPC2.1(pHLeu-tHMG1-UPC2.1);高拷贝的基因模块BTS1/ERG20,SaGGPSsyn.(pHHis-ERG20/BTS1-SaGGPSSyn.)得到的工程菌株ZD-T-018,产量为参照菌株ZD-T-000的2.7倍,达11.39mg/L,见图6。
实施例12、酿酒酵母基因工程菌ZD-T-021,ZD-T-022,ZD-T-023,ZD-T-024的构建(抗生素型)
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pLKanMX-tHMG1-UPC2.1。筛选培养的培养基为:1%Yeast Extract(酵母膏),2%Peptone(蛋白胨),2%Dextrose(葡萄糖),200mg/L G418;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-021。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pHKanMX-tHMG1-UPC2.1。筛选培养的培养基为:1%Yeast Extract(酵母膏),2%Peptone(蛋白胨),2%Dextrose(葡萄糖),200mg/L G418;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-022。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pLhphNT1-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:1%Yeast Extract(酵母膏),2%Peptone(蛋白胨),2%Dextrose(葡萄糖),300mg/L潮霉素;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-023。
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,分别转入质粒pHhphNT1-ERG20/BTS1-SaGGPSSyn.。筛选培养的培养基为:1%Yeast Extract(酵母膏),2%Peptone(蛋白胨),2%Dextrose(葡萄糖),300mg/L潮霉素;筛选培养的条件为:30度,培养36h以上。PCR鉴定出正确的阳性克隆,命名为菌株ZD-T-024。
实施例13、酿酒酵母基因工程菌ZD-T-025,ZD-T-026,ZD-T-027,ZD-T-028的构建(抗生素型)
采用和实施例4中相同的方法进行ZD-T-010感受态细胞的制备及转化,转入质粒pLKanMX-tHMG1-UPC2.1和pLhphN
生产次丹参酮二烯的酿酒酵母基因工程菌及其构建方法与应用专利购买费用说明
![]()
Q:办理专利转让的流程及所需资料
A:专利权人变更需要办理著录项目变更手续,有代理机构的,变更手续应当由代理机构办理。
1:专利变更应当使用专利局统一制作的“著录项目变更申报书”提出。
2:按规定缴纳著录项目变更手续费。
3:同时提交相关证明文件原件。
4:专利权转移的,变更后的专利权人委托新专利代理机构的,应当提交变更后的全体专利申请人签字或者盖章的委托书。
Q:专利著录项目变更费用如何缴交
A:(1)直接到国家知识产权局受理大厅收费窗口缴纳,(2)通过代办处缴纳,(3)通过邮局或者银行汇款,更多缴纳方式
Q:专利转让变更,多久能出结果
A:著录项目变更请求书递交后,一般1-2个月左右就会收到通知,国家知识产权局会下达《转让手续合格通知书》。











动态评分
0.0